BedokFunland JC's A Level H2 Chemistry Qns (Part 2)
-
Originally posted by Ephemeral:
8. (pg 204 reaction II) why do we need to deprotonate NH2OH first? Can reaction be carried out without deprotonation?
9. Is there a mistake in the mechanism in fig 7.9. ? Is the nucleophilic addition - elimination reaction and mechanism in syllabus?
10. (pg 207 tip 4) Is it due to hydride shift?
11. (pg 215) Does all phenyl containing tertiary alcohol form benzoic acid under prolonged heating with acidified KmO4? (pg 214) Do all tertiary alcohol without benzene ring form ketone and carboxylic acid? Do all pri (with K2Cr2O7) and sec alcohol with benzene ring still form aldehyde and ketone when oxidised?
12. (pg 228- 229 example 7.17) For synthesis of 2-methylprop-2-enal, isomer II is used (answer stated isomer III). Why is 2-methylprop-2-enal formed at the last step? It is minor product according to Zaitsev's rule (right?). Or does the presence of C=O result in 2-methylprop-2-enal being the major product?
Q8. No, deprotonation is necessary for this reaction because hydroxylamine (and all amines) are too weak a nucleophile to attack the ester (which is a weak electrophile because the partial positive charge on the C atom is only due to induction, not resonance ; resonance is the same reason why the weak nucleophile 2,4-DNPH only reacts with the more electrophilic aldehyde and ketone, but not the less electrophilic carboxylic acids, esters and amides).Q9. Yes, there's an error in the mechanism. While it's not required for the basic H2 syllabus, but if you're serious about getting your A grade, it'll be good for you to understand and be familiar with such mechanisms (not blindly memorize, of course).
For A grade H2 Chem, H3 Chem and Olympiad Chem students, here is the SOCl2 mechanism by James Ashenhurst : http://www.masterorganicchemistry.com/2011/12/03/reagent-friday-thionyl-chloride-socl2
For A grade H2 Chem, H3 Chem and Olympiad Chem students who are seriously intending to study Chemistry, Chemical Engineering, Biochemistry, Pharmacy, Dentistry or Medicine in the University, here is a more advanced discussion on the stereochemistry of the SOCl2 mechanism :

Image source : https://en.wikipedia.org/wiki/SNi
If you have difficulty understanding the diagram above, James Ashenhurst explains the reaction in detail here : http://www.masterorganicchemistry.com/2014/02/10/socl2-and-the-sni-mechanism/Q10. Yes, carbocation rearrangements can be due to hydride or alkyl shifts. See Leah Fisch's YouTube video here : https://www.youtube.com/watch?v=cSW9LDxtuoA, or Khan Academy's YouTube video here : https://www.khanacademy.org/science/organic-chemistry/substitution-elimination-reactions/e1-e2-tutorial/v/carbocations-and-rearrangements
Q11. Yes, and yes. The condition for side-chain oxidation by KMnO4 is that at least 1 benzylic H atom or benzylic OH group must be present.
Q12. Eh look carefully lah, there's only 1 possible dehydration (ie. elimination of H2O) product, as the 2 terminal or beta methyl groups are equivalent.
Also, when it comes to Markovnikov's or Zaitsev's rules, don't blindly apply or quote them. If Cambridge asks why such-&-such is the major product, you'll need to explain *why*, ie. the basis for *how* and *why* Markovnikov or Zaitsev's rule works (or in more challenging qns, may fail to work).
Good that you can suggest suspecting the C=O group, but such groups undermine Markovnikov's rule, not Zaitsev's rule, so it's irrelevant to this particular question. For an example of how the major product can controlled to be the Hofmann product instead of the Zaitsev product, see https://en.wikipedia.org/wiki/Zaitsev%27s_rule#Steric_effects.
-
Originally posted by Ephemeral:
for my Q12, I was wondering why the dehydration cannot occur with the H from the aldehyde (taking H from a C that is 'less rich' in H).
Why is the reactivity of halogenating agents as such: F>Cl>Br>I ?
I'll deal with your halogenating qn 1st :Your qn is ambiguous, do you refer to halogenation via free radical mechanism, or via nucleophilic (aliphatic or acyl) substitution, or via electrophilic aromatic substitution, etc? Never be ambiguous in your A level exam answers, if Cambridge asks "what is this reaction?" you should specify "halogenation via _________ (mechanism)".
So what exactly are you asking? Provide a context if relevant, eg. a specific TYS or prelim paper qn, or a website link, or a page number from a CS Toh book, or George Chong book, or Chan Kim Seng book, etc.
--------------------------------------------------------
Edited : "for my Q12, I was wondering why the dehydration cannot occur with the H from the aldehyde (taking H from a C that is 'less rich' in H)."
Eh sial lah! From your statement above, it shows you're just blindly memorizing and applying Markovnikov's and/or Zaitsev's rules without a proper understanding of *how* or *why* they usually work (and thus when they sometimes won't work)! You (ie. most Singapore JC students) jialat liao lah, this is the kind of teaching in Singapore JCs which illustrates the superiority of my BedokFunland JC pedagogy : *Understanding* then applying (as opposed to "blindly memorizing then applying" as taught in Singapore JCs).
As to your Zaitsev qn : Ahhh ok, what you mean is you're thinking of generating the ketene functional group from the acyloin functional group, well you could have just said so ;Þ
There are a couple of problems with your proposed reaction. First of all, it's not even thermodynamically feasible to generate a ketene (which is highly unstable and reactive) from an alpha-haloaldehyde/ketone, much less from an alpha-hydroxyaldehyde/ketone (ie. the acyloin functional group).
The dehydration of alcohols to alkene involves the the E2 mechanism (pri & sec alcohols) or E1 mechanism (sec & tert alcohols) catalyzed by an acid (see Jim Clark and Khan Academy and James Ashenhurst).
But when you have an acyloin functional group, it's more kinetically and thermodynamically feasible to protonate the carbonyl group rather than the hydroxy group, in which case hydrolysis of the aldehyde into the germinal diol occurs instead, which is unstable and simply dehydrates back into the aldehyde.
But since it's not entirely impossible or inconceivable (just less kinetically and thermodynamically favourable) for your proposed mechanism pathway to occur (ie. to generate the ketene functional group from the acyloin functional group) as a minor pathway or product via the E2 mechanism (E1 is even less feasible because of the destabilizing inter-nuclei repulsions between the partial and formal positively charged adjacent C atoms in the hypothetical carbocation intermediate), what happens then?
Under the reaction conditions to dehydrate an alcohol into an alkene (ie. concentrated H2SO4 acid catalyst), the highly unstable and reactive ketene product generated would be immediately acid-hydrolyzed into a carboxylic acid (try drawing out the mechanism for this reaction ; Cambridge asked data-based questions about the reaction of ketenes in both the UK / CIE and Singapore A level papers in recent years).
So the bottomline : good that you've considered the possibility* of obtaining a ketene as an alternative product, but unless the question gives you hints that the product is a ketene, you should stick to the more syllabus-familiar product of a normal alpha-carbonylalkene, rather than the more unstable ketene (H2 Chem students are expected to be able to deduce its instability by its structure). But nonetheless (as I said, good that you've thought-out-of-the-box* and considered the ketene product), in the A level exams, if you're not sure if the unusual product (here, the ketene) is a viable alternative product (or even one required by the Cambridge qn), then the exam-smart BedokFunland JC thing to do, is to give both answers (ie. draw structures of both alternative products), with labels or annotations as to which is likely to be the more stable (ie. thermodynamic) product, and hence major product.
* updated : actually it turned out you were just blindly applying Markovnikov's / Zaitsev's rule, not creatively thinking out-of-the-box... *sigh*
-
Originally posted by hoay:
In BF3 Boron atom has 6e can we call it an electrophile?
Secondly why LiAlH4 is more reactive than NaBH4 as reducing agent.
Yes, the BF3 molecule is an electrophilic Lewis acid (with the B atom being the dative bond acceptor). The term "Lewis acid" refers to its propensity to accept dative bonds, while the term "electrophile" refers to its propensity to seek electrons, so both terms are closely related, but not technically the same. In addition, the terms "nucleophile" and "electrophile" are by convention, favored for use specifically in Organic Chemistry, while the terms "Lewis base" and "Lewis acid" are overarching archetypes across all branches of Chemistry. For Cambridge A level Chemistry purposes, exam-smart students are advised to combine both terms (ie. any and all relevant terms or keywords for all questions in general) to secure the marks, even if Cambridge is usually reasonable and will accept either term.In case you were asking *why* BF3 is an electrophilic Lewis acid, it's because the B atom only has 3 bond pairs + 0 lone pairs, and thus lacks a noble gas stable octet valence shell electron configuration, and thus has a propensity to accept a dative bond to achieve a stable octet electron configuration.
For 2 closely related reasons. Firstly, because Al has a larger atomic radius than B, and thus polarizability of the electron bond pairs of the H- hydride ions increase with increasing distance from the central 'cationic' atom, as predicted by Coloumb's law, and also due to increasing shielding effect from larger number of inner shell electrons. Secondly, the lattice dissociation enthalpy for the covalent bonds with the hydride ions becomes less endothermic (ie. bond weakens) as bond length increases from BH4- to AlH4-, due to less effective overlap of more diffused electron shell orbitals involved in forming the sigma bonds. For both reasons above, concordantly and consequently, LiAlH4 is a significantly more effective delivery agent of H- hydride ion nucleophiles, compared to NaBH4.
-
Originally posted by Ng.keebin:
When you treat nitrobenzene with Sn in conc HCl to form phenylamine, why do you still need to add NaOH after that?
Because in the presence of acidic conc HCl, the product is the protonated conjugate acid phenylammonium ion (which is water soluble due to ion - permanent dipole interactions and hence difficult to separate from the other species present), which must thus be deprotonated by excess NaOH(aq) to obtain the desired product phenylamine, which must then be extracted via steam distillation.The experimental details of steam distillation are beyond A levels, but Cambridge can (and have in past A level papers) set challenging questions on the chemistry concepts underlying practical procedures (including steam distillation). Students seriously intending to score a distinction A grade for your A levels, are strongly advised to study the following webpages.
---------------------------------------------------------------------------------
Source : http://www.chemguide.co.uk/organicprops/aniline/preparation.html
Sodium hydroxide solution is added to the product of the first stage of the reaction. The phenylamine is formed together with a mixture of tetrahydroxozincate(II) and hexahydroxozincate(IV) complex tin ions formed from the tetrachlorozincate(II) and hexachlorozincate(IV) complex tin ions present during the acidic HCl stage. The phenylamine is finally separated from this mixture. The separation is long, tedious and potentially dangerous (and thus not experimentally carried out at A levels, but questions can still be asked on the procedure), involving steam distillation, solvent extraction and a final distillation.
---------------------------------------------------------------------------------
Source : http://chemistry.stackexchange.com/questions/10531/preparation-of-phenylamine-aniline
This is the procedure I (a Chemistry undergraduate student) carried out in the lab :
1.Reduction of nitrobenzene by using tin and HCl acid
2.After the completion of the reaction, NaOH added to neutralize excess acid and dissolve any Sn(OH)2(s)
3.Steam distillation is carried out to obtain suspension of phenylamine
4.Salting carried out where NaCl salt is added (called 'salting'). Aqueous layer discarded
5.Solvent extraction : dry ether added and mixture shaken vigorously. Aqueous layer discarded
6.Distillation under reduced pressure (ie. partial vacuum) carried out and solid phenylamine left in flask
My questions:
1. Instead of carrying out distillation under reduced pressure at the end of the procedure to remove the ether, could I have just carried out simple distillation?
2. Why is steam distillation suitable in part 3? Could I have used vacuum distillation here?
(See webpage for the answers to this Chem undergrad's qns).---------------------------------------------------------------------------------
Source : http://rod.beavon.org.uk/phenylamine_prep.htm
•What is the reaction between the tin and hydrochloric acid?
•Sn + 2HCl → Sn2+ + 2Cl- + H2
•What is the reducing agent, and to what is it oxidised?
•The reducing agent is Sn2+, which is oxidised to Sn4+.
•What is the initial precipitate, and what does it become on addition of excess sodium hydroxide?
•Tin(IV) hydroxide Sn(OH)4 (s). This is amphoteric, and with more alkali forms the soluble Sn(OH)6 2- hexahydroxozincate(IV) complex ion.
•Why is more water added to the solution?
•This enables a technique called steam distillation. In the large-scale preparation steam is blown through the mixture; in this one the steam is generated in situ. Phenylamine distils over with the steam; any unchanged nitrobenzene and the inorganic materials do not.
•Why is the initial distillate cloudy?
•It is a mixture of water and phenylamine which formas a cloudy emulsion
•What is the clear condensate?
•Water; by now all the phenylamine has distilled over.
•What is the purpose of adding sodium chloride?
•Phenylamine is significantly soluble in water, but very much less so in saturated sodium chloride solution. This process is called 'salting out'.
•What is the purpose of adding ethoxyethane?
•Solvent extraction; phenylamine is much more soluble in ethoxyethane than it is in water.
•Why are two portions of 4 cm3 used, rather than a single 8cm3 portion of of ethoxyethane?
•The theory of solvent extraction is a branch of chemical equilibria. It can be mathemically shown (and can be asked in data-based A level exam questions) that any solvent extraction is more effective if a given volume of extracting solvent is used in several portions rather than in a single one.
•Suggest a reason for using KOH as a drying agent, rather than the more conventional calcium chloride or sodium sulphate.
•The use of potassium hydroxide would also eliminate any traces of hydrochloric acid in the phenylamine.
•Why must all flames in the laboratory be extinguished?
•The vapour of ethoxyethane is very dense and will creep along bench-tops over a considerable distance. It is possible for an explosion to ensue even if the source of ignition is several metres away from where the ethoxyethane is being used.
•Why is the water run out of the condenser?
•Phenylamine has a sufficiently high boiling temperature for an air condenser to be efficient at condensing the vapour.
•Why is the phenylamine kept condensing on the thermometer bulb before allowing distillation to proceed?
•This allows the thermometer to come into thermal equilibrium with the vapour - a general necessity in distillation, but needing more care than usual if the boiling temperature of the distillate is high. Phenylamine boils at 184oC. -
Originally posted by hykel:
https://twitter.com/_hykelchem/status/707837536685588480?s=09
I don't really know how to answer both question 7 and 8. Please help!
For the HCJC qn, only C is possible, since the halogen radical only abstracts H atoms in propagation steps, leaving behind alkyl radicals which can form new C-C bonds with other alkyl radicals in termination steps. Options A, B and D require cleavage of C-C bonds, and/or other alkane molecules such as methane and ethane to be available. Try drawing out the reaction mechanisms, you'll find only C is possible.For the MJC qn, the 2 factors which determine product distribution during free radical substitution are : no. of H atoms substitutable, and stability of alkyl radical intermediates (aka reactivities of pri vs sec vs tert H atoms). Based on no. of H atoms substitutable, ratio of 1-bromo-2-methylpropane to 2-bromo-2-methylpropane should be 9 : 1. Based on stability of alkyl radical intermediates, ratio of 1-bromo-2-methylpropane to 2-bromo-2-methylpropane should be 1 : 6. Hence, actual ratio obtained (ie. combining the 2 factors mathematically by multiplication) = 9 x 1 : 1 x 6 = 9 : 6 = 3 : 2
-
Originally posted by Flying grenade:
Hi sir Ultima, may i ask,
a Lewis diagram = Dot and Dot structure
a Kekule diagram = Line and Dot structure
a Dot and Cross diagram = Dot and Cross structure
Many people, including teachers in Singapore JCs, are confused about the difference between these terms, and often use the term "Lewis structure" when they mean "Kekule structure"; Cambridge uses the term "Displayed Structural Formula".
For 'A' level purposes, Cambridge will accept any of the above, or even hybrid variants between.
a Lewis diagram = Dot and Dot structure
What dyou mean by Dot and Dot?
Thank you !
Lewis structures are dot-dot diagrams :
https://www.nyu.edu/classes/tuckerman/adv.chem/lectures/lecture_11/node1.htmlKekule structures are bond-line diagrams :
https://www.google.com.sg/search?source=hp&q=kekule+structure
http://chemwiki.ucdavis.edu/Core/Organic_Chemistry/Case_Studies/What_is_Organic%3F
Singapore JC teachers often wrongly call Kekule structures as Lewis structures. But it doesn't really matter, coz Cambridge doesn't use either term, rather, Cambridge will call it "displayed structural formula" or "full structural formula" (ie. the Kekule structure, with lone pairs not required to be shown unless the molecular or ionic geometry is also required), as opposed to dot-&-cross structures.
Lastly, Singapore JC teachers also wrongly call the resonance contributor of benzene as "the Kekule structure of benzene". (Actually here, it's not totally the Singapore JC teachers' error, becoz in a way it was Kekule's own error when he drew out the structure of benzene over a hundred years ago, but don't blame him, he was still the first human to come closest to the correct structure of benzene (you should check out the alternative proposed structures of benzene by other rival chemists), and back then, humans didn't understand the concept of resonance contributors and resonance hybrids (which Singapore JCs only properly teach to H3 Chem & OIympiad Chem students, depriving poor H2 Chem students of a proper understanding of Chemistry), so Mr Kekule was still the best man with the best idea back then).
This (what Singapore JCs call as the "Kekule structure of benzene") is more correctly called the Kekule/Lewis structure and skeletal structural formula (respectively) for the resonance contributor of benzene :
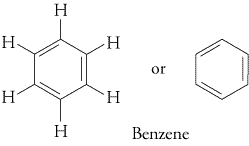
http://preparatorychemistry.com/Bishop_Resonance.htmThis (what Singapore JCs call as the "correct structure of benzene") is more correctly called the Kekule/Lewis structure and skeletal structural formula for the resonance hybrid of benzene :

http://www.tutorvista.com/chemistry/physical-properties-of-benzene
http://chemistrywithelevens.blogspot.sg/2012/05/rings-and-cyclones.htmlLastly, note that Cambridge will accept the skeletal formula for benzene even if the question specifies "full or displayed structural formula". And when drawing structures or mechanisms, Cambridge will accept either the resonance hybrid (ie. with the circle in the middle, which Singapore JCs anally insist you draw this instead of the resonance contributor with alternating double and single bonds, because they're afraid you JC H2 students are not intelligent enough to understand the intricacies of the differences between resonance contributor and resonance hybrid), or the resonance contributors (ie. with the alternating double and single bonds, which is actually the more correct structure to use when drawing reaction mechanisms, as you'll learn in the University, if you take Chemistry or Medicine in the University).
Cambridge will accept either the resonance contributor or hybrid, so no worries. Of course, the A level shortcut of using the resonance hybrid saves time, so go ahead and use it (most students can't even complete doing all questions in the A level paper during the A level exams). But if you want a deeper understanding of Chemistry, and also for some of the more challenging exam A grade distinction questions (eg. one of my BedokFunland JC H2 Chemistry questions : Under certain conditions, para- aminophenol, also known as para- hydroxyphenylamine, can be oxidized to generate a product with molecular formula C6H5NO. By drawing the simplified free radical mechanism to represent the reaction, elucidate the structure of the product.), you'll find that using the resonance contributor is actually superior to using the resonance hybrid.
-
Originally posted by Flying grenade:
ACJC/2014/P3/QN1(a)
I made a careless mistake doing this qn, i was required to draw dot and cross for N2O4, but i carelessly drew N2O2
However i realised that the dot and cross was good to draw with, in the sense all atoms in this molecule got full octet
https://www.dropbox.com/s/0dtmulqzsnxdj24/20160318_010332.jpg?dl=0
(Update, i just realised that drawing suits for Hydrazine, N2H4, maybe thats why N2O2 do not exist?)
However I'm surprised N2O2 doesn't exist in nature(or does it) !?
https://en.m.wikipedia.org/wiki/Nitrogen_oxide
No N2O2 in this wiki page
https://www.dropbox.com/s/o3hnr80cqvm29ah/Screenshot_2016-03-18-01-03-49.png?dl=0
Why doesnt N2O2 exist?
N2O2 exists, but under standard conditions at 25 deg C, it decomposes into NO monomers with thermodynamically favorable positive entropy change, with the position of equilibrium lying so far towards the NO monomer side that the molarity of N2O2 dimer is negligible. You need to cool NO into its liquid state (−152 °C) to have a significant molarity of N2O2 dimer. -
Originally posted by Flying grenade:
I was under the impression that dimer can only be formed by a dative bond, from one lone pair of an atom to the empty orbital of another atom.
Doesnt seem like N2O2 has a dative bond
Can help further elucidate /check my drawing above?
Thank you Ultima
Formation of dimers needn't necessarily involve dative covalent bonds lah, depending on the exact species involved, dimers can formed via hydrogen bonds or normal (ie. non-dative) covalent bond, especially for free radicals (they dimerize in a free-radical termination step, while dissociation into monomers is a free-radical initiation step).Ur drawing of N2O2 is fine, now draw the electron-flow curved-arrow reaction-mechanism for both the forward (free-radical termination) reaction to generate the N2O2 dimer, as well as the backward (free-radical initiation) reaction to dissociate the N2O2 dimer into NO monomers.
A very similar question was asked by Cambridge a couple of years ago, but many students couldn't do it, coz many Singapore JCs didn't teach their students how to properly draw the curved arrow mechanism for termination steps (most schools just teach their students to write the mechanism, not draw it with curved arrows).
-
Originally posted by Flying grenade:
AJC/2014/P3/Q1 (b)
Qn : BeCl2 has properties similar to those of aluminium chloride, AlCl3. Write a balanced equation to show the reaction of beryllium chloride with limited amount of water.
Answer : BeCl2 + H2O -> BeO + HCl
Or BeCl2 + 2H2O -> Be(OH)2 + 2HCl
But
AlCl3 + 6H2O -> [Al(H2O)6]3+ + 3Cl-
[Al(H2O)5(OH)]2+ 《》[Al(H2O)5(OH)]2+ +H+
How to solve this qn/ train of thought?
First thing first is it detect the phrase 'limited water'?
Yah, it's becoz of limited water, so hydrolysis of BeCl2(s) goes only so far as to generate BeO(s) or Be(OH)2(s), Cambridge will accept either equation, unless otherwise specified by the question. To get acidic solution like AlCl3(aq) or MgCl2(aq) via proton dissociation from water ligands, you need excess water :[Be(H2O)4]2+(aq) + 2Cl-(aq) + H2O(l) --> [Be(OH)(H2O)3]+(aq) + 2Cl-(aq) + H3O+(aq)
-
Originally posted by Flying grenade:
Can give example of H-bond dimers??
Can give examples of (normal) covalent bonded dimers? N2O4?
Yah, N2O4 is a dimer of NO2, and NO2 is a monomer of N2O4. The terms monomer and dimer are relationship adjectives, and have no meaning on its own. So "N2O4 is a dimer" is a wrong statement, but "N2O4 is a dimer of NO2 monomers" is a correct statement.For the most commonly asked at A levels H-bond dimer, click on the link below, followed by clicking on "images".
-
Originally posted by hykel:
https://twitter.com/_hykelchem/status/710759882488549376?s=09
I'm not sure how to get compound A and B. Please help :)
Step 3 is nucleophilic acyl substitution (addition-elimination) to get amide group. Step 4 is nucleophilic aliphatic substitution (SN2) to get ether group.In step 6, thionyl chloride converts carboxylic acid group to acyl chloride group.
For A grade H2 Chem, H3 Chem and Olympiad Chem students, here is the SOCl2 mechanism by James Ashenhurst : http://www.masterorganicchemistry.com/2011/12/03/reagent-friday-thionyl-chloride-socl2
For A grade H2 Chem, H3 Chem and Olympiad Chem students who are seriously intending to study Chemistry, Chemical Engineering, Biochemistry, Pharmacy, Dentistry or Medicine in the University, here is a more advanced discussion on the stereochemistry of the SOCl2 mechanism :
Image source : https://en.wikipedia.org/wiki/SNi
If you have difficulty understanding the diagram above, James Ashenhurst explains the reaction in detail here : http://www.masterorganicchemistry.com/2014/02/10/socl2-and-the-sni-mechanism/ -
Originally posted by Flying grenade:
Having said that, there is a significant difference between CO2 and XeF2, which is : in CO2, the central partially positively charged C atom has no lone pairs, while the central partially positively charged Xe atom has 3 lone pairs. Therefore the (relative) magnitude of permanent dipole - permanent dipole attractions will be expected to be more significant for CO2 than for XeF2. And yet XeF2 has a much higher melting point than CO2, because XeF2 has a lot more electrons, has much larger molecular size, and therefore has significantly more polarizable electron charge clouds.)
Why CO2's Pd-Pd attraction is relatively stronger than XeF2's Pd-Pd attraction??
I thought XeF2 would have a larger net dipole moment than CO2, hence more significant Pd-Pd than CO2??
Both XeF2 and CO2 are linear, and hence have no overall net dipole. However, because C has no lone pairs, it is more electron-deficient compared to Xe with 3 lone pairs. Hence, based on this factor alone, you might expect C to be more partial positive compared to Xe. However, as I pointed out years ago, the most important factor remains polarizability of electron charge clouds, and Xe being in Period 5, has a much larger atomic radius than C, as well as greater shielding effect from greater number of inner shell electrons, and as such, coupled with the greater electronegativity of F over O, the magnitude of partial positive charge on Xe will still be greater than on C. Consequently, the intermolecular van der Waals interactions (all 3 types of van der Waals forces : permanent dipole - permanent dipole Keesom forces, permanent dipole - induced dipole Debye forces, and instantaneous dipole - induced dipole London dispersion forces) will still be far stronger between XeF2 molecules compared to between CO2 molecules.Based on the simplified (and often erroneous) teachings of Singapore JCs, almost all H2 Chem students (and even some JC teachers) will be instantly dismissive of the idea of any permanent dipole - permanent dipole intermolecular interactions between (overall) non-polar molecules like CO2. And yet permanent dipole - permanent dipole Keesom forces *does* exist between some non-polar molecules like CO2, On a related note, during hydration which occurs before hydrolysis, (overall) non-polar CO2 can actually even participate in hydrogen bonding. I won't elaborate or explain why here on the forum (where's the fun in that?), interested students can go figure out for themselves (or ask their school teachers or private tutors).
-
Originally posted by Flying grenade:
When do we write Pd-Id force, i.e. force between a permanent dipole and a corresponding induced dipole (Debye force) ?
How to tell there's Pd-Id intermolecular attractions?
Sch didn't teach Pd-Id,
But i remember you teaching keesom-debye-vandawaals forces
The relative magnitude of pdpd attractions are affected by the number of lone pairs on the central atom and also the magnitude of partial positive on the central atom ah?
"in CO2, the central partially positively charged C atom has no lone pairs, while the central partially positively charged Xe atom has 3 lone pairs. Therefore the (relative) magnitude of permanent dipole - permanent dipole attractions will be expected to be more significant for CO2 than for XeF2."
*Cries* , i feel that there's so much that idontknow
Intermolecular permanent dipole - induced dipole Debye van der Waals forces exists between the polar part of a molecule (eg. polar carbonyl group of a large ketone), and the non-polar part of a neighboring (but same species) molecule (eg. non-polar alkyl group of a large ketone). This is why all 3 types of van der Waals forces usually exist between most molecules (and many Singapore JCs erroneously teach their students that dipole - dipole interactions isn't the same as van der Waals interactions, and in doing so mislead and confuse their students ; in fact all 3 types of dipole - dipole interactions *are* the 3 types of van der Waals forces).By definition of electronegativity (which Cambridge can test you in the Singapore A levels), electronegativity only affects bond pairs, not lone pairs. Hence, no matter how electronegative F is, only the electron density of the bond pairs will be polarized towards F. The 3 lone pairs on the Xe atom remains unpolarized.
-
Originally posted by Ephemeral:
Hello! Keebin and I did the organic chem deductive elucidation killer Q13 and we got a few answers as attached -
When O C4H6O6 is heated with concentrated sulfuric(VI) acid, two possible products M and G with molecular formulae C4H2O4 and C8H8O10 are formed as a result of intramolecular and intermolecular esterifications, respectively. Elucidate O, M, G.
https://twitter.com/123chem2016/status/711185220322463745
M4 is obtained frm O1 as well. Are they all possible? As for the G compounds, degree of unsaturation for G3 is 7 instead of 5. Technically G1 can also under elimination (of water) to form another C=C...
Benzaldehyde is less reactive than methanal. Why is that so? Why does delocalisation of electrons through resonance reduce the electrophilicity of carbonyl carbon and make it less reactive? If the the electrons are delocalised over to the O atom, then doesn't it increase the partial positive charge on carbonyl C and hence make it more susceptible to nucleophilic addition?
When Grignard reagents react with carbonyl, a covalent bond is formed between O and MgX. Why can't it be an ionic bond?
All your structures are wrong. The question specified the products are generated as a result of intra and inter molecular esterifications, and dehydration of alcohol to alkene is not involved (as verified by the degree of unsaturation). O1 (hence M1 and G1 as well) is/are wrong because geminal diols spontaneously dehydrate to generate aldehydes / ketones, with a negligible molarity of the geminal diol present at equilibrium (it's true that KMnO4 and K2Cr2O7 oxidizes aldehydes via their hydrolysis into geminal diols, but KMnO4 and K2Cr2O7 are able to thusly shift the position of equilibrium to the RHS as the geminal diol is oxidized into carboxylic acid). Your O2 (and thus M2, G2, M3, G3 as well) is/are wrong because your M2, M3 and G3 suffers from excessive ring strain due to angle strain (an approximately 90 deg 4-membered ring is already strained from sp3 C atoms ideally 109.5 deg, let alone sp2 vinylic C atoms ideally 120 deg C). Now that I've revealed your structures of O are wrong, there's only 1 possible structure left, which is the correct structure of O, and hence the correct M and G follows accordingly.Because of both sterics and electronics : the electrophilic C atom in benzaldehyde suffers from greater steric hindrance (for the incoming nucleophile) compared to methanol ; in terms of electronics methanal has a valid resonance contributor in which the carbonyl C atom has a unipositive formal charge, while in benzaldehyde the unipositive formal charge is delocalized from the benzylic C atom over to the ortho, para and ortho C atoms in the benzene ring, consequently in their resonance hybrids, the magnitude of partial positive charge and hence electrophilicity, is significantly greater in the methanal C atom than in the benzaldehyde C atom.
The O-Mg bond in the intermediate R-O-Mg-X upon nucleophilic addition of the Grignard reagent, has significant covalent and ionic character both. Unless otherwise specified by the question, Cambridge will accept it whether you draw & describe the bond as ionic (with significant covalent character), or as covalent (with significant ionic character). It's actually better to draw it as ionic (ie. O- +Mg with formal charges between them), to show Cambridge that you're aware of the greater magnitude of electronegativity difference between O & Mg compared to C & Mg (which is more covalent than ionic).
-
Originally posted by Flying grenade:
[Al(H2O)6]3+ is formed from AlCl3 in excess water, because Al is in period 3, there's energetically accessible vacant d-orbitals , so Al can expand it's octet, is it?
Why doesnt it form [Al(H2O)4]3+?
[Be(H2O)4]2+ is formed instead of [Be(H2O)6]2+ from BeCl2 in excess water, because period 2 elements are unable to expand their octet is it?
Yes correct. Even if Al can accept just 4 H2O ligands, but it'll prefer 6, because bond forming (including dative bonds) are exothermic and hence thermodynamically favorable.
Originally posted by Flying grenade:Hi Ultima, may i ask you,
AJC /2014/P3/2(b)
State the shape and bond angle of [Pt(NH3)4]2+
I think the challenge to this question is, to determine the number of valence electrons of centre atom Pt right?
This is what i did : i do not know how to determine valence e- of Pt, as we did not learn how to write electronic configuration beyond period 3 (is it?) .
So i just assume Pt has 2 valence e- , so lost 2 valence e- , and 4 NH3 ligands datively bonded to Pt, i get a tetrahedral shape, bond angle 109°
The answer wrote :
Shape : square planar
Bond angle : 90°
But, accept tetrahedral 109°
How did they get sq planar or assumed pt has 6 valence e-?
Did a google search for electronic config of Pt , it's complex and their still debating abt it
May i ask u for guidance for the thought process towards this qn?
For transition metal cations, you can ignore any lone pairs from the metal atom itself, and deduce its geometry solely based on the coordinate dative bonds accepted from its ligands. If the ligands are huge, eg. Cl- ligands, then to avoid excessive steric strain from van der Waals repulsions between the ligands, there will only be 4 instead of 6 ligands. When you know there are 4 ligands, then whether the geometry of the coordination complex is tetrahedral or square planar depends on the Jahn–Teller effect (which is of course way beyond the A level H2 syllabus), as well as other conditions that favor either stereoisomer (some coordination complexes can exist as an equilibrium between tetrahedral and square planar, with the position of equilibrium affected by other factors beyond A levels).For A level purposes, unless the coordination complex is well-known and student familiarity is expected (eg. such as Tetraamminecopper(II) sulfate and Cisplatin), Cambridge (and AJC here) will accept either square planar or tetrahedral, as long as the corresponding bond angles are correct.
Originally posted by Flying grenade:Thank you.
Yeah it's perplexing, AJ wrote the answer square planar, and then a second statement below
also accept tetrahedral,
Its like suggesting sq planar should come to mind first rather than tetrahedral.
But after your input, i feel that tetrahedral is more straightforward.
I was thinking, is it becos they just assume structure consisting of 4 bond pairs? If they did this, then there's a possibility of seesaw/distorted tetrahedral.
Why then, distorted tetrahedral not accepted/wrong?
Or is it acceptable?
As I said, ignore lone pairs from the transition metal itself, take it that the only electron charge clouds present are the dative bonds from the ligands. And the AJC qn was closely related to Cisplatin, which is a famous chemotherapy drug and hence you should be aware that it's square planar and has cis-trans isomerism (although it's not in the Singapore H2 syllabus, but it's in the IB and other A level Chem syllabuses, and if you're serious about A grade, you should expose yourself to stuff like this beyond your own A level syllabus). -
Originally posted by Flying grenade:
Ultima, everytime u take the A levels, your paper 1 - 5 all 100% correct?
Buddha said, "If I don't go to Hell, who will go to Hell?"BedokFunland JC said, "If I don't get A grade, who will get A grade?"
Because I know how Cambridge marks, I know the distinction A grade boundary and I know how the bell-curve works, hence I can choose to skip whichever questions or practical procedures I'm not interested in doing, and still get my A grade every year. So I don't get 100% full marks because I deliberately skip many questions or practical procedures, but the questions that I choose to do will be 99.9% correct, give or take a couple of silly careless mistakes (you don't actually need to outrun the bear, you only need to outrun the other guy who is also being chased by the bear).
-
Originally posted by Flying grenade:
Hi Ultima, may i ask you,
I know the priority of the functional groups for aliphatic molecules as shown here :
https://www.dropbox.com/s/25ncr8p5i5y1e9x/20150107_153638-1.jpg?dl=0
However may i ask about the priority of the functional groups for aromatic molecules?
I was confused about this :
https://en.m.wikipedia.org/wiki/2-Nitrotoluene
Its called 2nitromethylbenzene here, i.e. toluene has higher priority?
Compared to https://en.m.wikipedia.org/wiki/Chlorotoluene, 1-chloro-2-methylbenzene , i.e. chlorine has higher priority here?
As far as professional chemists are concerned, so-called priorities of functional groups in naming organic molecules (in contrast, Cahn–Ingold–Prelog priority rules are clear, logical and must be consistently followed for uniformity and unambiguity's sake) are merely IUPAC guidelines (not strict rules), what's far more important (and critical) is that the species is unambiguously identified. Which means there are multiple correct or acceptable names for most organic molecules. For instance, look at the "Other names" below the "IUPAC name" for 2-Nitrotoluene here : https://en.wikipedia.org/wiki/2-Nitrotoluene. All these "other names" are also correct, IUPAC is a just a guideline.For A level purposes, Cambridge too, allows for flexibility in naming as long as it is unambiguous. And on a more relevant and practical note for JC students, is that Cambridge doesn't test naming much at A levels, the question can give a complex name for an organic molecule and the student must be able to draw it out, but not vice-versa.
As a general rule (remember Chemistry, like real-life, must be intelligent and sensible, not dogmatic and meaningless), instead of memorizing priorities from your school lecture notes, just ask yourself, which functional group is more chemically reactive (ie. the most nucleophilic, electrophilic, lower Ea, etc), then such a functional group should be highlighted first in the name (because the most reactive functional group defines the molecule's most important properties), and hence give it priority. Do this because it makes intelligent sense to do so, not because your school lecture notes dogmatically say so.
PS. Just like how you (ie. everyone reading this) are advised to always be an intelligent, free-thinking individual who is self-responsible for what you choose to believe, be the captain of your own soul, instead of blindly following religious dogma of any religion.
-
Originally posted by Flying grenade:
Ultima, may i ask you how to determine if a (covalent) molecule can form favourable (ion-dipole) interactions with water ,hence soluble?
E.g https://www.dropbox.com/s/mb6a48ty02rgc7v/20160323_145224.jpg?dl=0
This is a covalent organic molecule, hence cannot use Energy of dissolution=Hydration enthalpy - lattice dissociation energy right?
Is it look at partial positive and partial negative?
Is it that, even if a molecule can form favourable (ion dipole) interactions with water, but not 100% always soluble?
Molecules are not ions, hence cannot have ion - permanent dipole interactions with protic polar water.For A level purposes, Solution enthalpy / entropy = Lattice dissociation enthalpy / entropy + Hydration enthalpy / entropy, is specifically for ionic species. (While it can theoretically be applied to covalent species, but due to the varying nature of different covalent species and their lack of a lattice structure under standard conditions, the relevant data becomes difficult to obtain and infeasible to apply with this Hess law derived formula, so instead direct experimental techniques are used instead).
Look at polarity and proticity. Molecule P is non-ionic (ie. no positive for negative formal charges present), aprotic, and relatively non-polar, hence is insoluble in water.
Yes, for instance, Cambridge can ask why amino acids are least water-soluble in their zwitterionic form at isoelectric point.
-
Originally posted by Flying grenade:
A protein has its lowest solubility at its isoelectric point. At isoelectric point, the amino acid carries no net electrical charge. Without a net charge, protein-protein interactions and precipitation are more likely, rather than to interact with water and undergo solvation.
Can get full marks?
Zero marks. The website you quoted from failed to explain *why* precipitation rather than solvation (when the solvent is water, better to specify hydration) occurs at isoelectric point.I won't reveal the correct answer here, JC students can go ask your school teacher or private tutor. FlyingGrenade, no doubt you're gonna keep googling for the answer, but don't post the answer here and ruin the self-learning experience for others.
-
Originally posted by hoay:
Cyanohydrins can be made from carbonyl compounds by generating CN– ions from HCN in the presence of a weak base.
In a similar reaction, –CH2CO2CH3 ions are generated from CH3CO2CH3 by strong bases.
Which compound can be made from an aldehyde and CH3CO2CH3 in the presence of a strong base?A CH3CH(OH)CO2CH3
B CH3CO2CH2CH(OH)CH3
C CH3CH2CH(OH)CH2CO2CH3
D (CH3)2C(OH)CH2CO2CH3
A and D are ruled out... as A does not use the correct reagent while D is a ketone
B and C are close. Both B and C are aldehyde...B is ethanal and in C it is propanal.
It's just the orientation otherwise both B and C seems correct..
Please help.
Hi Hoay,Yes, this is the classic Cambridge "ester group condensed structural formula" trick question. C is the answer. B is wrong because when the condensed structural formula of an ester is read from left to right as R1COOR2, the same ester read from right to left must be written as R2OCOR1 (and not R2COOR1). Hence, B would be correct only if it is changed from CH3COOCH2CH(OH)CH3 to CH3OCOCH2CH(OH)CH3.
Bonus related question (for A grade H2 Chem students) :
Draw the relevant resonance contributors and resonance hybrid of the ester's conjugate base to explain why that particular proton is the most acidic and is hence abstracted from the ester in the presence of a non-nucleophilic and strong Bronsted-Lowry base, eg. lithium diisopropylamide. -
Originally posted by hoay:
dont you think that in the presence of strong base –CH2CO2CH3 ions will attack either ethanal to produce the compund in choice B. It does make sense that this will be formed...then why we treat it as ester.
The nucleophile here, is the conjugate base of the ester. As such, the product of nucleophilic addition (of the conjugate base of the ester unto the aldehyde electrophile) will naturally include the condensed structural formula of the ester, and hence my previous post about how the ester is written from left to right, versus from right to left, is relevant in differentiating between options B and C. -
Originally posted by Flying grenade:
Hi Ultima , may i ask you,
Markovnikoff's rule states that in the addition of H-X or H-OH to a C=C bond in an asymmetrical alkene, the H atom attaches itself to a C atom that already holds the greater amount of Hydrogen atoms.
May i ask this : https://www.dropbox.com/s/9bahzdn6n96o91s/20160324_223727.jpg?dl=0
How to know Br and OH attach where??
For electrophilic addition of X2 (aq) to (asymmetrical) alkene to form halohydrin , how to determine X and OH form where?
Cambridge will *not* accept Markovnikov's rule (or Zaitsev's rule, for that matter) as a reason, if Cambridge asks why a particular product is the major product (eg. "Draw the major product, and explain why it is the major product."). To get full marks, you need to draw both mechanisms (or at the very least, both the carbocation intermediates) for both products, then specify which carbocation is the more stable carbocation (ie. benzylic carbocation > allylic carbocation > tertiary carbocation > secondary carbocation > primary carbocation), then explain "The more stable the intermediate, the lower the Ea required, hence the faster the rate of reaction, hence we get more of the product, which we label as the major product."
Originally posted by Flying grenade:Since O is more electronegative than Br, is it O will form at the benzylic C atom ?
First of all, nothing "forms" at the benzylic C atom, wrong choice of words.Secondly, your reasoning is totally wrong (ie. it's not about electronegativity of O vs Br, because even if it was fluorine used instead of bromine, and F is of course more electronegative than O, the major and minor products discussed above will still be similar, albeit with *additional* possible beyond-H2-syllabus products due to the strongly oxidizing and highly reactive nature of fluorine).
Draw the full mechanisms to get both major & minor products (actually, there'll be a total of 3 possible products excluding stereochemistry when using aqueous halogen, can you figure out why?), and upload the image.
Note : many Singapore JCs teach wrongly that OH- attacks the carbocation, but it is actually H2O that is the competing nucleophile. Don't make this error in your mechanism.
-
Originally posted by MightyBiscuits:
Oh sry i didnt notice.
https://www.instagram.com/p/BDYfruSSTTz/?taken-by=mightybiscuits1 (qns 19 ans)
https://www.instagram.com/p/BDYfnpkSTTl/?taken-by=mightybiscuits1 (qns 19)
I dont understand what is the meaning of hydrolysed. Does it mean that it is water of crystallisation? And whats the difference between hydrogen chloride and hydrochloric acid.
Ok, so your own queries isn't actually about CS Toh's worked solution, which you understand perfectly?Hydrolysis (noun) or hydrolyze (verb) or hydrolyzed (adjective), refers to chemical reaction with water. If you ask your school teacher or other private tutors or websites, a lot of them will tell you it means 'water-splitting' (hydro-lyze), but it could be either water or the other reactant, or both that is being lyzed or split, so "chemical reaction with water" is actually a superior definition, in terms of chemistry concepts. In this question, germanium chloride reacts with water to generate germanium oxide.
Hydrogen chloride refers to HCl(g) or HCl(l), while hydrochloric acid refers to HCl(aq), which is actually hydroxonium chloride, ie. H3O+(aq) and Cl-(aq).
-
^_^
-
Originally posted by Flying grenade:
Hi ultima, may i ask what other factors might affect this other than steric hindrance?
https://www.dropbox.com/s/2l6bxfaaarpk3wu/20160330_180801.jpg?dl=0
Qn part d)
Ultima, may i ask u if cstoh chemistry study guide have steric hindrance? Tried finding ,flipped every page at the org chem section, cant find, index doesn't have steric hindrance also.
May i ask what do you mean by electronics?
Tertiary alkyl halides (such as in ur qn part d), undergo SN1, and primary alkyl halides undergo SN2, for 2 reasons : sterics and electronics.Sterics and electronics are also the reasons why aldehydes are more electrophilic than ketones, so if you only have 1 mole of a nucleophile reacting with 1 mole of an electrophile with both aldehyde and ketone groups present, it is the aldehyde, not the ketone, which will be the electrophilic group accepting the nucleophile.
Electronics refers to the relative magnitudes and distribution of charges (both formal charges in resonance contributors and partial charges in resonance hybrids) as a consequence of either induction or resonance or hyperconjugation (the 1st 2 of which are more important for the H2 syllabus).
{kind=link}