BedokFunland JC's A Level H2 Chemistry Qns (Part 2)
-
Originally posted by Audi:
Just one simple question... so simple that it bugs me when I can't determine which is the preferred answer.
Alright, so we were asked how we can distinguish between I2 and Br2.
There were 2 options I find quite Ok
First is reaction with sodium thoiosulfate. This seems reasonable because we know that I- ions will become I2 and Br- becomes NaBr. If I am not wrong, NaBr should be white (as is all group I ionic compounds?) and I2 gives a yellow solution
Second option is using aq Fe2+ then add aq NaOH.
we know I2 does not react witih Fe2+... so Br2 WILL, giving Fe3+ and Br -. NaOH and react with Fe3+ to give Fe(OH)3 ppt?
I chose the first answer anyway... was the answer that just "came" to me at that time. Any other take? Thanks
Method 1 :
Assuming standard molarities, thiosulfate will indeed reduce both iodine and bromine, to iodide and bromide respectively, and is itself oxidized to tetrathionate and sulfate(VI) respectively. All of which are (equally) colourless.Method 2 :
Assuming standard molarities, calculating cell potential (ie. reduction potential at cathode + oxidation potential at anode) will show that only one halogen is a sufficiently strong oxidizing agent to oxidize Fe2+ to Fe3+. Thereafter, adding OH- will precipitate Fe(OH)2(s) and Fe(OH)3(s) in the two separate test solutions, which have distinct colours of dark green and reddish brown respectively. -
^_^
-
Q1. The species C6H5CH2CH(CH3)Cl undergoes an intramolecular reaction (with a Lewis acid catalyst). What is the name for this reaction?
Answer :
In regards to the benzene ring nucleophile, the reaction is an electrophilic aromatic substitution.
In regards to the alkyl chloride electrophile, the reaction is a SN1 nucleophilic substitution.
Considering both the nucleophile and electrophile, the reaction is known as a Friedel-Crafts alkylation of the benzene ring.
Although the electrophile is a secondary alkyl halide, and can theoretically undergo either SN1 or SN2 reaction, however because the benzene ring is a weak nucleophile (since its pi electrons are delocalized by resonance (through the sideways overlapping of p orbitals), and are thus less available for nucleophilic attack), an SN2 mechanism which requires a strong nucleophile, is therefore not possible.
And since a Lewis acid catalyst is used, a (secondary) carbocation electrophile intermediate is generated (that is readily attacked by the benzene ring nucleophile), and hence this electrophilic aromatic substitution reaction proceeds via an SN1 nucleophilic substitution mechanism, and the overall reaction is known as a Friedel-Crafts alkylation of the benzene ring.
Q2. The species C6H5CH2CH2COCl undergoes an intramolecular reaction (with or without a Lewis acid catalyst). What is the name for this reaction?
Answer :
In regards to the benzene ring nucleophile, the reaction is an electrophilic aromatic substitution.
In regards to the acyl chloride electrophile, the reaction is a nucleophilic acyl substitution.
If a Lewis acid catalyst is not used, the nucleophilic substitution on the acyl halide electrophile proceeds via an addition-elimination mechanism. (In industrial practice, a Lewis acid catalyst is always used to lower activation energy and speed up the reaction, because time = money.)
If a Lewis acid catalyst is used, a (resonance stabilized) acylium cation electrophile intermediate is generated (that is readily attacked by the benzene ring nucleophile), and hence this Lewis acid catalyzed electrophilic aromatic substitution - cum - nucleophilic acyl substitution reaction is known overall as a Friedel-Crafts acylation of the benzene ring.
-
Originally posted by Audi:
1. 20.0cm3 of 0.10 mol dm-3 aq Na2CO3 was titrated witih 0.20 mol dm-3 aqueous HCl.
The 2 base hydrolysis constants of the carbonate ion are Kb1 = 2.08 X 10^-3 mol dm-3
Kb2 = 2.32 X 10^-8 mol dm-3
Calculate pH of solution when 0cm3, 5cm3, 15cm3,20cm3 and 25 cm3 of HCl was added.
Sketch the titration curve labeling the points and indicating buffer regions.
Do ICF (moles) tables for the different volumes of NaOH(aq) added. You'll find that 0cm3 and 5cm3 are before the 1st equivalence point, while 15cm3 is between 1st and 2nd equivalence points, and 20cm3 is for 2nd equivalence point, and 25cm3 is past the 2nd equivalence point.To find pH at these various points :
0cm3 : Use Kb of CO3 2-
5cm3 : Use Henderson-Hasselbalch with [CO3 2-] / [HCO3 -]
15cm3 : Use Henderson-Hasselbalch with [HCO3 -] / [H2CO3]
20cm3 : Use Ka of H2CO3. Be mindful of the volume change.
25cm3 : Calculate [H+] contributed from HCl only, and thus calculate pH. Write a statement to explain that because HCl is a strong acid and H2CO3 is a weak acid, the protons dissociated from HCl suppresses the proton dissociation from the weak acid H2CO3 (as predicted by Le Chatelier's principle), and thus we only need to consider [H+] from HCl. Be mindful of the volume change.
-
A buffer solution is titrated with NaOH(aq). Upon addition of 3 volumes of NaOH(aq), the buffer solution becomes most effective. Equivalence point is attained when 8 volumes of NaOH(aq) is added.
a) Calculate the ratio of the molarities of both members of the conjugate acid-base pair of the buffer solution, before the NaOH(aq) is added.
b) Given that the original pH of the buffer solution (ie. before NaOH(aq) is added) is 4.16, calculate the proton dissociation constant of the acidic component of the buffer.
Solution :
a) At maximum buffer capacity :HA + OH- ---> A- + H2O
Initial (mol) 8y 3y x-8y n.a.
Change (mol) -3y -3y +3y n.a
Final (mol) 5y 0 x-5y n.a.At equivalence point :
HA + OH- ---> A- + H2O
Initial (mol) 8y 8y x-8y n.a.
Change (mol) -8y -8y +8y n.a.
Final (mol) 0 0 x n.a.First, complete the ICF table for equivalence point. Next, complete the ICF table for maximum buffer capacity, bearing in mind that whether for maximum buffer capacity or equivalence point, the initial moles of HA and A- are the same.
Since at maximum buffer capacity, [HA] = [A-], this implies 5y = x-5y and hence x = 10y.
Substituting x = 10y into the initial moles of A-, we have moles of A- = 2y.
Therefore, the ratio of the amounts of both members of the conjugate acid-base pair present in the buffer (before NaOH(aq) is added), is 8y HA : 2y A-, ie. 4 HA : 1 A-
b) Using the Henderson-Hasselbalch equation, we have
pH = pKa + log ( [base] / [acid] )
4.16 = pKa + log (1/4)
pKa = 4.762
Ka = 1.73 x 10^-5
-
^_^
-
kickme asked :
Give the organic products of the reaction of (i) ethanal and (ii) propanone with,
a)LiAlH4 followed by dilute H2SO4
I understand that LiAlH4 implies reduction but what does H2SO4 do?b) H2N-OH
Is the nucleophile OH-? It is possible for NH2+ to be the nucleophile instead?Just a question: is it true that any compound with NH2+ can be used as brady's reagent?
Visualize the mechanism :
The H- guy (nucleophile) from LiAlH4 shoots out its balls (lone pair) to attack the chio (delta +ve) girl (electrophilic), the pi bond shifts up to O (to avoid violating C's octet), and you get the alkoxide (ie. alkyl oxide) intermediate. The H2SO4 protonates the alkoxide ion intermediate to generate the final product : the alcohol.-----------------------------------
In hydroxylamine, both the N and the O atoms are nucleophilic centers, and for some reactions you'll get a mixture of products. Generally though, the N atom is more nucleophilic, because of two reasons :
N is less elecronegative than O, and thus more willing to 'shoot out its balls' (ie. lone pair) to nucleophilically attack the 'girl' (ie. electrophile). Furthermore, the N atom is bonded to two (less electronegative and thus electron donating) H atoms, in contrast to only one H atom for the O atom.
-----------------------------------
Visualize the mechanism : the N atom attacks the C atom, the pi bond shifts up, proton transfer (from N to O), 2nd protonation of O to generate good leaving group H2O+, N gets its balls back becomes horny again, attacks the C atom 2nd time, H2O+ eliminated as H2O, N loses proton to lost its +ve formal charge and regains its balls (lone pair) again.
If the OH group wasn't present, the product would be an imine (C=N functional group). Since the OH group remains bonded to the N atom, the final product with the (RRC=N-OH) functional group is known as an oxime :
http://en.wikipedia.org/wiki/Oxime-----------------------------------
No, Brady's reagent refers specifically to 2,4-Dinitrophenylhydrazine. The two nitro groups are electron-withdrawing by both induction and resonance, and hence weakens the balls (electron density) of the benzene and amine nucleophile. Being a highly weakened, sterically hindered (benzene ring is large and bulky) nucleophile is important, because recal that 2,4-DNPH reacts only with very chio or feminine (electrophilic) electrophiles like aldehydes and ketones, but not with less chio or feminine (electrophilic) electrophiles like carboxylic acids, esters and amides, even though these compounds also contain the carbonyl C=O) group.
The reason for the reduced electrophilicity of the carboxylic acids, esters and amides, relates to resonance. In aldehydes and ketones, when the C=O pi bond shifts up, the alpha C atoms lack the balls (lone pair) and thus cannot donate electrons by resonance to stabilize the carbocation resonance contributor. In contrast, in carboxylic acids, esters and amides, the adjacent O or N atom can donate a lone pair by resonance to form a pi bond with the carbonyl carbon, preventing a +ve formal charge. Hence in the resonance hybrid, the carbonyl C atom of carboxylic acids, esters and amides are less chio, feminine and electrophilic, and won't be attacked by the weak balls guy (weakened nucleophile) the 2,4-DNPH.
Generally, the more stabilized (eg. by resonance, by induction, by hydrogen bonding, etc) you are, the less reactive you are. Carboxylic acids, esters and amides are stabilized by resonance, aldehydes and ketones are not.
--------------------------------------
kickme replied :
Thank you, for part (a) is it true that all reduction reaction of aldehydes and ketones to generate alcohols requires an acidified medium?
For part (b), I didn't quite understand the mechanism... I only understood "N atom attacks the C atom, the pi bond shifts up, proton transfer (from N to O)," Is the mechanism nucleophilic addition?
No, it must be LiAlH4 (in dry ether) followed by protonation (eg. via hydrolysis).
Yes, the mechanism is nucleophilic addition for the 1st step only. Google or Wikipedia out the mechanism to form imines and enamines. -
Is this ___________________ in the syllabus?
If you wanna boost your probability of scoring a distinction grade and stand victorious atop all the dead bodies on the bell-curve, then you should have the will and courage to boldly go where no H2 student hath gone before...
-
blacktoast94 asked :
Hi all! does anyone know how to do this?
partial hydrolysis of a polypeptide with 2 different enzymes, A and B, produced the following peptides. deduce the order in which the amino acids are bonded together in the original polypeptide.
enzyme A:
try-lys-gly
leu-ala
arg-try-his-meth
arg-gly
enzyme B:gly-arg-try
try-lys
ala-arg-gly
his-meth-leucan explain how to go about doing such questions too? my lecture notes don't explain :/ . thank you! :)
There is no chemistry formula for such questions. Treat it as a jigsaw logic puzzle. Just copy-and-paste the smaller bits (ie. the oligopeptides) to form the larger picture (ie. the polypeptide). Once done, confirm your answer by checking that all the oligopeptides are indeed all present in the correct sequence in your hypothesized polypeptide.
Based on the work of both enzymes, the polypeptide (ie. the answer) is :
try-lys-gly-arg-try-his-meth-leu-ala-arg-glyenzyme A:
try-lys-gly (confirmed)
leu-ala (confirmed)
arg-try-his-meth (confirmed)
arg-gly (confirmed)enzyme B:
gly-arg-try (confirmed)
try-lys (confirmed)
ala-arg-gly (confirmed)
his-meth-leu (confirmed)Note that the required assumption (which may or may not be stated or asked by the question) is that the convention for writing peptide sequences (for all oligopeptides and the polypeptide) is to put the N-terminus on the left and write the sequence from N- to C-terminus.
http://en.wikipedia.org/wiki/N-terminus
http://en.wikipedia.org/wiki/C-terminus -
blacktoast94 wrote :
Hi ultima thank you for your reply! :D i'm really grateful that you are able to help me all the time ^_^ .
i also have another question: i'm quite confused about alpihatic alcohols. are they neutral or basic? carboxylic acid and alcohol can undergo acid-base reaction, yet, according to what i've learnt for organic the aliphatic alcohols are neutral or even v. v. slightly acidic
so which one should i use if the alchohol is an aqueous solution? :qmarkblackface:You're totally welcome blacktoast :xizao::qmarkstickout:, and also welcome to continue asking your questions here... countdown : only 8 months left to 2012 'A' levels! :biggrins:
As Einstein said, everything is relative. Alcohols are relatively neutral, meaning a solution of an aliphatic alcohol has a pH of 7 at rtp. But everything (including alcohols and water) can function as a base (if placed in a strongly acidic environment) or as an acid (if placed in a strongly basic environment).
Say you wanted to protonate your alcohol. Simply add concentrated strong acids eg. H2SO4. Once protonated, the OH group becomes OH2+ which is a good leaving group, allowing for nucleophilic substitution to occur readily.
Say you wanted to deprotonate your alcohol. Simply add sodium metal. This is both a redox reaction (since Na is oxidized to Na+, and H+ from alcohol is reduced to H2 gas) and a Bronsted-Lowry acid-base proton transfer reaction (since the alcohol is deprotonated to generate its conjugate base, the alkoxide ion).
You're expected to know, as part of the H2 syllabus, all three points described above : that alcohols are relatively neutral (compared to more acidic organic species such as phenol, and compared to more basic species such as amines; and explain all of these by electronegativity argument, inductive effects, resonance delocalization, etc), but you must also know how to use alcohol as an acid (ie. deprotonate it) and how to use alcohol as a base (ie. protonate it).
Countdown to 2012 'A' Levels! :biggrins: -
Metformin is an anti-diabetic, anti-cancer drug, that has proven effective in the treament and even prevention, of both diseases. It is available here in Singapore, but requires a doctor's prescription (so if you intend to take Metformin to prevent diabetes or cancer, you still need to ask your doctor for a prescription first).
Draw the mechanism for the synthesis of Metformin. (Hint : it's a simple two-step mechanism : nucleophilic addition, followed by (intramolecular) proton transfer reaction).

-
kickme asked :
Distinguish between CH2COOCH3 and HCOOCH2CH3 using a chemical test.
I see that both are esters. But i dont see any test which i can use to distinguish between the 2 esters. One of them is acidic and the other is not. Thats all i can see...Solution :
Heat under reflux with acidified KMnO4.
Upon acidic hydrolysis (since you're heating under acidic conditions) to generate carboxylic acids and alcohols, followed by oxidation, the ester HCOOCH2CH3 will yield methanoic acid, which would be further oxidized to which will be further oxidized to carbonic(IV) acid, which exists in equilibrium with, and hence can decompose into, carbon dioxide gas (gaseous products will leave the reactant mixture, pulling the position of equilibrium over to the right, as predicted by Le Chatelier's principle) and water.
It's ironic that "kickme" didn't know the answer to this (admittedly fairly challenging for JC students) question, but he knew something else that 99% of most JC students didn't : that one ester is acidic and the other is not.
CH2COOCH3 is acidic because when deprotonation occurs with (ie. the alpha proton is abstracted away by) a strong base (eg. sodium metal Na, or sodium amide NaNH2), the conjugate base -CHCOOCH3 can be stabilized by having its negative charge delocalized by resonance over to the electronegative O atom of the carbonyl ester group. This is not possible for the other ester, HCOOCH2CH3.
-
Source :
http://www.titrations.info/back-titration1.435 g sample of dry CaCO3 and CaCl2 mixture was dissolved in 25.00 mL of 0.9892 M HCl solution. What was CaCl2 percentage in original sample, if 21.48 mL of 0.09312 M NaOH was used to titrate excess HCl?
Solution :
During titration 21.48×0.09312=2.000 mmole HCl was neutralized. Initially there was 25.00×0.9892=24.73 mmole of HCl used, so during CaCO3 dissolution 24.73-2.000=22.73 mmole of acid reacted. As calcium carbonate reacts with hydrochloric acid 1:2 (2 moles of acid per 1 mole of carbonate), original sample contained 22.73/2=11.37 mmole of CaCO3, or 1.137 g (assuming molar mass of CaCO3 is 100.0 g). So original sample contained 1.137/1.435×100%=79.27% CaCO3 and 100.0-72.27%=20.73% CaCl2.Direct complexometric Al3+ determination is difficult, as Al3+ reacts with EDTA very slowly. To solution containing some unknown amount of Al3+ cations 50.00 mL of 0.05000 M EDTA solution was added. After 30 minutes excess EDTA was titrated with 0.04875 M Zn2+ solution. What was the amount of Al3+ if 17.58 mL of titrant was used?
Solution :
EDTA reacts with both Al3+ and Zn2+ in 1:1 ratio. There was 50.00×0.05000=2.500 mmole of EDTA used, and 17.58×0.04875=0.8569 mmole was found to be left. Thus there was 2.500-0.8569=1.643 mmole of Al3+ in the sample.
-
^_^
-
^_^
-
Originally posted by H4x0ru5:
Ah thank you very much. Really enlighten me about this.
I have a last question relating to ions.
Iodine Tetrafluoride, IF4 - is a anion.
Iodine, the central atom, has 4 bond pairs and 2 lone pairs. The Fluorine atom has 1 bond pair, 3 lone pairs.
This gives Iodine a formal charge of -1. Each fluorine atom has a charge of 0.
Why doesn't the compound IF4 exist, since without the extra electron attached to Iodine, Iodine would have a 4 bond pairs, and 3 electrons attached to it. The formal charge of Iodine would therefore be 7-3-(8/2) = 0 Why does it exists as an anion instead of a compound ?
For it to exist as a molecule (ie. ionic charge = zero), one or more atoms (to be precise, the I atom) would have an unpaired electron (to be precise, 1.5 lone pairs, and the ion would be a free radical) and is thus too unstable to exist.
IF4-
The -ve formal charge is on the central I atom (since it has a larger atomic radius and a lower charge density and is thus more stable, despite its lower electronegativity).
In addition, note that F (being in period 2) cannot expand its octet, and thus to have a stable octet, it must have 1 bond pair and 3 lone pairs, and hence no formal charge (being in Grp VII).
Since I is in Grp VII, to have a -ve formal charge, we deduce that it has 8 valence electrons, ie. 4 bond pairs, 2 lone pairs.
Hence the electron geometry is octahedral, and the ionic geometry is square planar.
-
Originally posted by Jetihyun:
25.0 cm3 of a solution of M2O5 of concentration 0.100moldm-3 is reduced by sulphur dioxide to a lower oxidation state. To re-oxidise M to its original oxidation number, this required 50.00cm3 of 0.0200moldm-3 potassium manganate (VII) in acidified medium. To what oxidation number was M reduced to by sulfur dioxide?
I am really confused by the question. How do I use the mol ratio to find the oxidation number. I can't solve similar questions due to this problem too....

Thanks in advance

Write reduction half-equation of MnO4- to Mn2+.MnO4– + 8H+ + 5e– � Mn2+ + 4H2O
Find moles of MnO4- required.
50.00cm3 of 0.0200moldm-3 = 1 x 10^-3 mol
Hence find moles of electrons transferred.
1 x 10^-3 x 5 = 5 x 10^-3 mol
Find moles of M2O5 present.
25.0 cm3 of 0.100moldm-3 = 2.5 x 10^-3 mol
Find moles of M atoms present.
2.5 x 10^-3 x 2 = 5 x 10^-3
Hence find moles of electrons transferred per mole of M atoms.
(5 x 10^-3) / (5 x 10^-3) = 1
Find OS of M in M2O5.
+5
Hence find reduced OS of M.
(+5) + (-1) = +4
-
kickme asked :
Suggest what must be done to the orchid before it can be an electrode in the electroplating process and name the substance used for the other electrode.
My answer is to soak the entire orchid in Au3+ and the other electrode should be made of Au.
Am i right?Yes, the anode should be made of gold. The flower however, should be coated with graphite powder or paint.
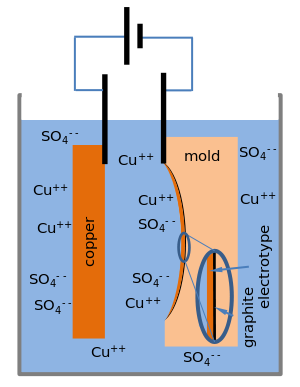
http://en.wikipedia.org/wiki/Electrotyping-----------------------------
kickme persisted :
Oh... Thanks. But wont the powder be washed off?
On another note, there are so many equation involving water in the data booklet. How do we know which is relevant to the reduction or oxidation of water?Nope, won't fall off.
Graphite powder is in the form of a sticky paste. Graphite paint is naturally sticky as well (like all other paints, otherwise the customer will demand a refund!).
For electrolytic cells, just use H2O itself as the reactant (for both oxidation and reduction equations).
For galvanic-voltaic cells, apply chemistry sense : if the solution is acidic, use reduction of H+ and oxidation of H2O; If the solution is alkaline, use reduction of H2O and oxidation of OH-. -
Originally posted by hoay:
So we can say that all the atoms in BF3 are co-planar while this is not the case in CH4?
Correct.Co-planar implies exactly planar, while peri-planar implies near or almost planar (eg. some bonds/atoms may deviate slightly due to van der Waals repulsion).
Both terms may be used interchangeably for 'A' level purposes.
-
kickme asked :
Why is it that the boiling point of CCl4 is greater than that of SiCl4?
Shouldn't SiCl4 have the higher boiling point given the greater electronegativity difference and larger molecular size?
2)Do ionic character in a covalent compound strengthen or weaken the bond?
3)Do covalent character in an ionic compound strengthen or weaken the bond?
Q1.
Silicon is a metalloid, and as such is significantly less electronegative compared to carbon. The Cl atoms in SiCl4 consequently have a larger magnitude of partial negative charge (compared to in CCl4), which results in a greater degree of van der Waals repulsion (here : lone-pair lone-pair repulsion). Hence SiCl4 has a lower boiling point compared to CCl4.Q2 & Q3.
Case by case basis (because depending on multiple factors such as charge densities and effectiveness of orbital overlaps, sometimes ionic bonds are stronger, sometimes covalent bonds are stronger).
Sometimes (eg. sodium versus silver halides), with greater covalent character, the (part-covalent-part-ionic) bond strength increases. Sometimes (eg. carbon versus silicon halide) the opposite is true.
For 'A' level purposes, use whichever explanation helps your case, or helps explain the data given in the question, or helps to answer the question.
However, what is more generally consistent (although there are always exceptions, eg. CCl4 versus SiCl4), is that the greater the covalent character, the lower the boiling point. -
Originally posted by Kahynickel:
CCl4 vs SiCl4
UltimaOnline posted:
The Cl atoms in SiCl4 consequently have a larger magnitude of partial negative charge (compared to in CCl4), which results in a greater degree of van der Waals repulsion (here : lone-pair lone-pair repulsion). Hence SiCl4 has a lower boiling point compared to CCl4.
Both molecules are symmetrical. Why are you not taking into account the larger vander Waal's forces in case of SiCl4 due to the larger number of elctrons as compared to CCl4?
Are you saying that the difference of just 6 more electrons in SiCl4 are not going to make it having a higher boiling point than CCl4.? Please explain.
Can you mention other similar cases?
In Chemistry just as in real-life, there are multiple factors in every situation, and commonly enough, some of these factors are contradictory.
Yes, the greater number of electrons and greater molecular size is indeed in favour of SiCl4 having stronger van der Waals over CCl4.
At the same time however, the greater magnitude of partial negative charges on the Cl atoms of SiCl4 is also in favour of CCl4 having stonger van der Waals over SiCl4.
So which outweighs which? We humans can argue all we want based on theoretial principles, but in the end, the Universe has already decided (by mathematics) which outweighs which, and we humans have to carry out experiments to find out what the Universe has already decided : in this context, experiments have proven that CCl4 has a higher boiling point over SiCl4, and therefore we humans can deduce that the second factor outweighs the first factor.
Another similar example, would be that of comparing the bond angles of NH3 versus NCl3. There are two contradictory factors in this regard :
Because the N-Cl bond lengths are significantly greater than N-H bond lengths, one might expect the inter-atomic-nuclei repulsions, and thus bond angles, to be smaller for NCl3 compared to for NH3.
But because Cl has a significantly larger atomic radius and has lone pairs, while H has a significantly smaller atomic radius and has no lone pairs, one might expect the inter-lone-pair repulsions, and thus bond angles, to be larger for NCl3 compared to for NH3.
So which factor wins? As it turns out, experimental evidence has shown that the bond angles of NCl3 and NH3 are almost identical, the former 107.1 deg and the latter 107.8 deg. And therefore we humans can deduce that both factors cancel each other out almost equally.
-
Explain why the C-C bond dissociation enthalpy in cyclobutane is significantly less endothermic compared to the C-C bond dissociation enthalpy given in the Data Booklet.
Solution :
Ring strain (ie. angle strain and steric strain) weakens the C-C bond. Molecules including C atoms with 4 bond pairs and 0 lone pairs, are most stable when the C atoms are sp3 hybridized with a bond angle of 109.5 deg. Cyclobutane's bond angles vary, depending on conformation, but all bond angles are significantly smaller than 109.5 deg, constituting a significant deviation (from the ideal tetrahedral bond angles) that results in significant angle strain (ie. inter-electron repulsion between bond pairs) and steric strain (ie. van der Waals repulsion between groups of atoms) which weakens the C-C bonds of cyclobutane (and all similarly small cycloalkanes).
-
Originally posted by Audi:
Ok, need some urgent help here.
How do I convert BrCH2CH2Br to HO2C-CO2H?
Thank you!
Heat under reflux with aqueous NaOH, followed by heating under reflux with acidified K2Cr2O7 (not KMnO4!). -
^_^
-
^_^