BedokFunland JC's A Level H2 Chemistry Qns (Part 2)
-
Kickme asked :
I was given the structure of Psilocin and was asked if the following statement is true. The nitrogen-containing group in the ring has a lower pKb than the nitrogen-containing group in the side chain.
UltimaOnline helpfully replied :Psilocin is a psychotropic drug that distorts the user's perception of time, similar to the fictional Slo-Mo drug featured in the currently screening movie (Judge) Dredd.

http://en.wikipedia.org/wiki/Psilocin
The secondary amine N atom on the ring is less basic, compared to the tertiary N atom in the side chain. This however, is not because a tertiary amine has an additional electron-donating by induction alkyl group (because in point of fact, in contrary to the erroneously simplified version taught in JCs, a tertiary amine is actually less basic compared to a secondary amine, but that is a completely separate discussion).The correct reason is because the lone pair of the cyclic secondary amine, is extensively delocalized away by resonance (made possible thanks to the sideways overlap of p orbitals of the N atom and the pi orbitals of both the alkene and the benzene ring), and consequently is less available to accept a proton.
This means the ring amine is a weaker base than that of the side-chain amine.
This means the Kb of the ring amine is smaller than that of the side-chain amine.
This means the pKb of the ring amine is larger than that of the side-chain amine.
-----------------------------------------------------
Memo felt perplexed :Wait... The diagram (From wikipedia)
Both N atom are sp3 hybridised. Is it?
Not quite. If the lone pair on the N atom is delocalized by resonance, then the lone pair resides in an unhybridized p orbital, which means the N atom is sp2 hybridized, ie. 120 degrees, using it's hybridized orbital to form its sigma bonds. -
KickMe pleaded :
The alpha -COOH group and the side chain -COOH groups' pKa seem to be out of line with theory. Presence of additional -NH2 group, being electron donating should result in the alpha -COOH group being less acidic since it destabalises the carboxylate ion formed. However, the alpha -COOH group has a pKa value which is lower than the side chain -COOH group. How do I account for this observation?
UltimaOnline came to the rescue :
The reason why you (and many JC students) have this misconception problem, is because you (ie. many JCs fail to teach you to) differentiate between induction versus resonance.
NH2 group may be electron donating by resonance (if applicable), but it is electron withdrawing by induction (since N is more electronegative than C, and even moreso electron-withdrawing when the N atom has a positive formal charge in a protonated NH3+ group).
In alpha-aminocarboxylic acids, only the electron-withdrawing inductive effect is applicable, and not the electron-donating resonance effect, since in alpha-aminocarboxylic acids, you do not have a pi orbital (eg. such as found in alkenes and benzene) available to overlap with the unhybridized p orbital of the N atom, which is a prerequisite for resonance delocalization to occur.
KickMe was enlightened :
I see! Is it true that the inductive effect takes place all the time if resonance doesn't occur? Another question I have is, in a 2-substituted benzene ring, sometimes the substituent is electron donating and at other times, electron withdrawing due to differences in the position of substitution. How then do we determine if the substituent is electron donating by resonance or electron withdrawing by inductive effect?
UltimaOnline continued his illuminatory teachings :Yes.
Resonance effect usually outweighs inductive effect, with the exception of halogens whose electron-withdrawing by induction effect has been experimentally proven to outweigh its electron-donating by resonance effect.
If there is a lone pair on the atom outside the benzene ring, then the substituent is able to donate electrons by resonance. Thus the substituent is consequently ortho-para directing (because the ortho and para positional isomers have a particularly stable resonance contributor in which every atom has a stable octet), and also an activator (unless the substituent is a halogen atom), meaning that the substituent increases the nucleophilicity of the benzene ring, activating it.
If the atom outside the benzene ring has a formal or partial positive charge (eg. in the nitro group and the carboxylic acid group respectively), it is electron withdrawing by induction, and is consequently a meta director (because the ortho and para positional isomers have a particularly unstable resonance contributor in which the positive formal charged C atom is directly adjacent to the positive partial charged C atom) and also a deactivator, meaning that the substituent decreases the nucleophilicity of the benzene ring, deactivating it. -
kickme asked :
Drawing PO4 3- dot and cross diagram
When drawing this ion, how do I know if I should give more than one electron to the same oxygen atom when allocating the 3- charge?
I would also like to confirm if the structure for NO3- ion, is one with a single bond, a double bond and a dative covalent bond.
I tried googling but the structures I obtained were probably too realistic and beyond the 'A' levels knowledge.Yes, your described structure suffices for (a resonance contributor of) the nitrate ion.
For the phosphate ion, the trinegative ionic charge is the sum of 3 individual uninegative formal charges, 1 uninegative formal charge on each of the 3 singly bonded O atoms (which therefore have 3 lone pairs and 1 bond pair). There will be a doubly bonded O atom (with 2 lone pairs and 2 bond pairs) which therefore has no formal charge.
Similarly, the resonance hybrid is the sum average of the resonance contributors. The nitrate and phosphate ions are stabilized by having their negative charge(s) delocalized by resonance over the electronegative O atoms. Ideally, students should be able to appreciate and draw, both the resonance contributors and the resonance hybrids for all species.
Singapore JCs do not properly teach the concepts of formal charges and resonance to their H2 students (usually these are only taught to H3 and Olympiad students), but these two concepts (amongst others) are actually fundamental to a proper understanding of chemistry, and constitute the fundamental teachings for my BedokFunland JC's curriculum approach to exceling in H2 Chemistry by virtue of having an actual understanding, rather than blind memorizing, of Chemistry.kickme replied :
A question on the phosphate ion, according to your description, there will be 3 single bonds and 1 double bond around the P atom, thus the number of electrons surrounding the P atom is 10. Why is it that we do not replace the double bond with a dative covalent bond instead, allowing the P atom to achieve octet.
To elaborate further on your original qn, why "give more than one electron to the same oxygen atom when allocating the 3- charge" :
For an O atom to have a dinegative formal charge without violating it's octet configuration (since O is in period 2 and does not have vacant, energetically accessible 3d orbitals to accomodate an expanded octet), it would have to have 4 lone pairs, and 0 bond pairs, ie. the oxide O2- anion. Obviously, such a species can not be part of a polyatomic species, but stands alone as a monoatomic ion.
To address your new qn :
A dative covalent bond is really just a normal bond. The idea of 'dative' bonding is flawed and misguided. It is caused by physical and inorganic chemists being more lazy than organic chemists (specifically in this particular regard). The organic chemist's approach of not using the dative bond arrowhead symbol, is actually more correct. Afterall, a dative bond arrowhead is really nothing more than a mini-mechanism (for which an organic chemist would draw two stages, using curved arrows).
Next, assuming you draw your structure as a unipositive formal charged P atom singly bonded to 4 uninegatively formally charged O atoms (ie. your dative bonding version), that's fine as well, and is indeed one of the resonance contributors that contribute to the actual structure, termed as the resonance hybrid.
In other words, in the multiple resonance contributors possible, each O atom 'takes turn' to become doubly bonded with no formal charge. Hence in the resonance hybrid, the magnitude of negative charge on each O atom, is somewhere between 0 and 1-, and the central P atom bears a significant partial positive charge, due to both the induction effect and resonance effect.
Similarly, the fact that carbonyl C atom of aldehydes and ketones bear a significant partial positive charge and are hence significantly electrophile, is actually due to both induction effect and resonance effect, and not just because of induction effect, as many JCs oversimplify and erroneously teach to H2 Chem students.Lastly, you should always draw the Kekule structure first (what some JCs call the Lewis structure, and what Cambridge calls the displayed structural formula), and then redraw it as a dot-and-cross diagram. For dot-and-cross diagrams, do not indicate formal charges, because the positive formal charge looks like a cross. But as long as it is not a dot-and-cross diagram, you should always indicate all formal charges (either before or after dative bond formation, depending on whether metal atoms are present or not; if metal atoms are present, eg. in complex ions, then OS is more accurate than formal charges, and in such case OS is equivalent to formal charges before the dative bonds were formed, ie. negative formal charges on the ligands instead of the metal ion, since metals are electropositive and ligands are more electronegative).
Kickme posted :
Thanks, just a question about your last paragraph, what do you mean when you write: "you should always draw the Kekule structure first"? Do you mean that as an aid to drawing the dot and cross diagram or as a requirement by the question?
As an aid. Coz dot-and-cross structures are (quite frankly) childish, a remnant from the little days of the 'O' levels. We do not use dot-and-cross structures in the University. Hence, drawing Kekule structures (ie. displayed structural formulae) should become 2nd nature for all 'A' level students, and only if the exam question requires it, then you convert it (by drawing next to the Kekule structure) to a dot-and-cross diagram.
When converting Kekule structures to dot-and-cross diagrams, bear in mind the following :
1) A dative bond must be represented by either a dot-dot or cross-cross. Cambridge will penalize you for drawing a dative bond arrowhead in a dot-and-cross diagram (ie. the way you would do it for a Kekule structure).
2) An anion must have a hollow dot, to represent the additional electron gained from (what used to be) the cation, which is usually a metal.
For instance, in the nitrate(V) ion, NO3-, there are two singly bonded O atoms with a uninegative formal charge each. One of these singly bonded O atoms gained this uninegative formal charge because it accepted a dative bond (ie. dot-dot or cross-cross bond pair), while the other singly bonded O atom gained the uninegative formal charge from the loss of a proton (H+ ion) from it's conjugate acid form of HNO3, and it is this singly bonded (non-dative bond, so use a dot-cross for this bond) O atom that will contain a hollow dot in one of its lone pairs. The hollow dot represents an electron from the H atom, before the H+ ion dissociated.
In heterolytic bond cleavage, the bond pair becomes a lone pair on the more electronegative atom, which is why when a proton is transferred from an acid to a base, the curved arrows are from the base to the acid, and the proton's old bond pair becoming a lone pair on the O atom of the conjugate base (assuming it's an oxoacid). -
Kick me asked :
What is the shape of a complex with a central metal ion that has a coordination number 4? Is it square planar or tetrahedral? Eg. {CuCl4}2-
Some are tetrahedral, some are square planar. When in doubt, write "since there are 4 donor atoms (from 4 monodentate ligands), the complex ion is expected to have either a tetrahedral or square planar geometry".
For most cases, Cambridge will accept either.
However, for some complex ions, eg. tetraaminediaquocopper(II) ion, the student is expected to be aware that it is square planar rather than tetrahedral.
For some complex ions, eg. your aforementioned tetrachlorocuprate(II) ion, both geometries are possible and do exist, depending on other factors such as the identity of the counter ion :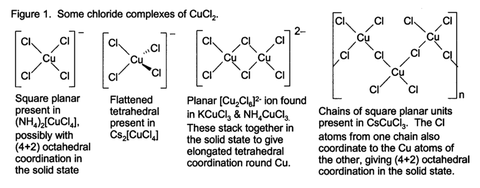
-
^_^
-
Kickme asked :
Is it chemically correct to write, "more branched structure"?
UltimaOnline replied :
It's even more chemically correct to write, "a more extensively branched isomeric structure".
Kickme asked :
Would the following statement to answer why a particular structure has a lower boiling point be correct?
A, having a more extensive branched isomeric structure as compared to B has a more spherical electron cloud which reduces the surface area for induced dipole-induced dipole(id-id) interactions, resulting in weaker id-id interactions which require less energy to overcome.UltimaOnline replied :
Yes that's fine, but the correct term required by Cambridge (don't trust Singapore JCs) is "instantaneous dipole - induced dipole", and the full term is "instantaneous dipole - induced dipole London dispersion van der Waals" forces or interactions.
Kickme asked :
Another question is, is it chemically correct to write 'breaking of a bond'?
UltimaOnline replied :
Yes that's fine, but specify covalent or ionic.
Did you know??? (ie. more stuff that Singapore JCs do not teach you)
There's a technical difference between "energy" and "enthalpy", but for 'A' level Chem purposes and for most reactions to 3 sig fig, these two terms may be used interchangeably.
Energy change is heat change under constant volume.
Enthalpy change is heat change under constant pressure.
For some reactions, there can be a significant difference between enthalpy change and energy change. For instance, in the exothermic reaction of reacting an acid with a metal carbonate, since some heat energy will be used up by the liberated CO2 to do work to push away atmospheric gases, enthalpy change will be slightly less exothermic compared to energy change.
Did you know???
There are actually two types of "lattice energy/enthalpy", one endothermic, one exothermic.
The energy/enthalpy associated with the breaking of ionic bonds is more correctly called "endothermic lattice dissociation energy/enthalpy".
The energy/enthalpy associated with the forming of ionic bonds is more correctly called "exothermic lattice formation energy/enthalpy".
There are actually two types of "covalent bond energy/enthalpy", one endothermic, one exothermic.
The energy/enthalpy associated with the breaking of covalent bonds is more correctly called "endothermic bond dissociation energy/enthalpy".
The energy/enthalpy associated with the forming of covalent bonds is more correctly called "exothermic bond formation energy/enthalpy".
Cambridge, by default, is referring to "endothermic bond dissociation energy/enthalpy" when asking about covalent "bond energy". Yet, when asking about ionic "lattice energy", Cambridge is referring to "exothermic lattice formation energy/enthalpy".
Which is silly (that the covalent one should by default be about bond breaking, while the ionic one should be by default about bond forming).
Which is why I always advise my BedokFunland JC and all other students who wanna be exam-smart, to always give both (eg. when asked for definitions, or in answering calculation questions), when the question is ambiguous.
Eg. After applying Hess Law, if you get the value of 100kJ/mol, write both :
"Applying Hess Law, we determine the lattice formation energy/enthalpy to be -100kJ/mol, and the lattice dissociation energy/enthalpy to be +100kJ/mol." -
[Singapore] - Millionaire Medical Doctor dying of Cancer at 40
Below is the transcript of the talk of Dr. Richard Teo, who is a 40-year-old millionaire and cosmetic surgeon with a stage-4 terminal lung cancer and came to share with the D1 class his life experience on 19-Jan-2012.
Hi good morning to all of you. My voice is a bit hoarse, so please bear with me. I thought I'll just introduce myself. My name is Richard, I'm a medical doctor. And I thought I'll
just share some thoughts of my life. It's my pleasure to be invited by prof. Hopefully, it can get you thinking about how... as you pursue this.. embarking on your training to become dental surgeons, to think about other things as well.Since young, I am a typical product of today's society. Relatively successful product that society requires.. From young, I came from a below average family. I was told by the media... and people around me that happiness is about success. And that success is about being wealthy. With this mind-set, I've always be extremely competitive, since I was young.
Not only do I need to go to the top school, I need to have success in all fields. Uniform groups, track, everything. I needed to get trophies, needed to be successful, I needed to have colours award, national colours award, everything. So I was highly competitive since young. I went on to medical school, graduated as a doctor. Some of you may know that within the medical faculty, ophthalmology is one of the most highly sought after specialities. So I went after that as well. I was given a traineeship in ophthalmology, I was also given a research scholarship by NUS to develop lasers to treat the eye.
So in the process, I was given 2 patents, one for the medical devices, and another for the lasers. And you know what, all this academic achievements did not bring me any wealth. So once I completed my bond with MOH, I decided that this is taking too long, the training in eye surgery is just taking too long. And there's lots of money to be made in the private sector. If you're aware, in the last few years, there is this rise in aesthetic medicine. Tons of money to be made there. So I decided, well, enough of staying in institution, it's time to leave. So I quit my training halfway and I went on to set up my aesthetic clinic... in town, together with a day surgery centre.
You know the irony is that people do not make heroes out average GP (general practitioner), family physicians. They don't. They make heroes out of people who are rich and famous. People who are not happy to pay $20 to see a GP, the same person have no qualms paying ten thousand dollars for a liposuction, 15 thousand dollars for a breast augmentation, and so on and so forth. So it's a no brainer isn't? Why do you want to be a gp? Become an aesthetic physician. So instead of healing the sick and ill, I decided that I'll become a glorified beautician. So, business was good, very good. It started off with waiting of one week, then became 3weeks, then one month, then 2 months, then 3 months. I was overwhelmed; there were just too many patients. Vanities are fantastic business. I employed one doctor, the second doctor, the 3rd doctor, the 4th doctor. And within the 1st year, we're already raking in millions. Just the 1st year. But never is enough because I was so obsessed with it. I started to expand into Indonesia to get all the rich Indonesian tai-tais who wouldn't blink an eye to have a procedure done. So life was really good.
So what do I do with the spare cash. How do I spend my weekends? Typically, I'll have car club gatherings. I take out my track car, with spare cash I got myself a track car. We have car club gatherings. We'll go up to Sepang in Malaysia. We'll go for car racing. And it was my life. With other spare cash, what do i do? I get myself a Ferrari. At that time, the 458 wasn't out, it's just a spider convertible, 430. This is a friend of mine, a schoolmate who is a forex trader, a banker. So he got a red one, he was wanting all along a red one, I was getting the silver one.
So what do I do after getting a car? It's time to buy a house, to build our own bungalows. So we go around looking for a land to build our own bungalows, we went around hunting. So how do i live my life? Well, we all think we have to mix around with the rich and famous. This is one of the Miss Universe. So we hang around with the beautiful, rich and famous. This by the way is an internet founder. So this is how we spend our lives, with dining and all the restaurants and Michelin Chefs you know.
So I reach a point in life that I got everything for my life. I was at the pinnacle of my career and all. That's me one year ago in the gym and I thought I was like, having everything under control and reaching the pinnacle.
Well, I was wrong. I didn't have everything under control. About last year March, I started to develop backache in the middle of nowhere. I thought maybe it was all the heavy squats I was doing. So I went to SGH, saw my classmate to do an MRI, to make sure it's not a slipped disc or anything. And that evening, he called me up and said that we found bone marrow replacement in your spine. I said, sorry what does that mean? I mean I know what it means, but I couldn't accept that. I was like “Are you serious?” I was still running around going to the gym you know. But we had more scans the next day, PET scans - positrons emission scans, they found that actually I have stage 4 terminal lung cancer. I was like "Whoa where did that come from?” It has already spread to the brain, the spine, the liver and the adrenals. And you know one moment I was there, totally thinking that I have everything under control, thinking that I've reached the pinnacle of my life. But the next moment, I have just lost it.
This is a CT scan of the lungs itself. If you look at it, every single dot there is a tumour. We call this miliaries tumour. And in fact, I have tens of thousands of them in the lungs. So, I was told that even with chemotherapy, that I'll have about 3-4months at most. Did my life come crushing on, of course it did, who wouldn't? I went into depression, of course, severe depression and I thought I had everything.
See the irony is that all these things that I have, the success, the trophies, my cars, my house and all. I thought that brought me happiness. But i was feeling really down, having severe depression. Having all these thoughts of my possessions, they brought me no joy. The thought of... You know, I can hug my Ferrari to sleep, no... No, it is not going to happen. It brought not a single comfort during my last ten months. And I thought they were, but they were not true happiness. But it wasn't. What really brought me joy in the last ten months was interaction with people, my loved ones, friends, people who genuinely care about me, they laugh and cry with me, and they are able to identify the pain and suffering I was going through. That brought joy to me, happiness. None of the things I have, all the possessions, and I thought those were supposed to bring me happiness. But it didn't, because if it did, I would have felt happy think about it, when I was feeling most down..
You know the classical Chinese New Year that is coming up. In the past, what do I do? Well, I will usually drive my flashy car to do my rounds, visit my relatives, to show it off to my friends. And I thought that was joy, you know. I thought that was really joy. But do you really think that my relatives and friends, whom some of them have difficulty trying to make ends meet, that will truly share the joy with me? Seeing me driving my flashy car and showing off to them? No, no way. They won’t be sharing joy with me. They were having problems trying to make ends meet, taking public transport. In fact i think, what I have done is more like you know, making them envious, jealous of all I have. In fact, sometimes even hatred.
Those are what we call objects of envy. I have them, I show them off to them and I feel it can fill my own pride and ego. That didn't bring any joy to these people, to my friends and relatives, and I thought they were real joy.
Well, let me just share another story with you. You know when I was about your age, I stayed in king Edward VII hall. I had this friend whom I thought was strange. Her name is Jennifer, we're still good friends. And as I walk along the path, she would, if she sees a snail, she would actually pick up the snail and put it along the grass patch. I was like why do you need to do that? Why dirty your hands? It’s just a snail. The truth is she could feel for the snail. The thought of being crushed to death is real to her, but to me it's just a snail. If you can't get out of the pathway of humans then you deserve to be crushed, it’s part of evolution isn't it? What an irony isn't it?
There I was being trained as a doctor, to be compassionate, to be able to empathise; but I couldn't. As a house officer, I graduated from medical school, posted to the oncology department at NUH. And, every day, every other day I witness death in the cancer department. When I see how they suffered, I see all the pain they went through. I see all the morphine they have to press every few minutes just to relieve their pain. I see them struggling with their oxygen breathing their last breath and all. But it was just a job. When I went to clinic every day, to the wards every day, take blood, give the medication but was the patient real to me? They weren't real to me. It was just a job, I do it, I get out of the ward, I can't wait to get home, I do my own stuff.
Was the pain, was the suffering the patients went through real? No. Of course I know all the medical terms to describe how they feel, all the suffering they went through. But in truth, I did not know how they feel, not until I became a patient. It is until now; I truly understand how they feel. And, if you ask me, would I have been a very different doctor if I were to re-live my life now, I can tell you yes I will. Because I truly understand how the patients feel now. And sometimes, you have to learn it the hard way.
Even as you start just your first year, and you embark this journey to become dental surgeons, let me just challenge you on two fronts.
Inevitably, all of you here will start to go into private practice. You will start to accumulate wealth. I can guarantee you. Just doing an implant can bring you thousands of dollars, it's fantastic money. And actually there is nothing wrong with being successful, with being rich or wealthy, absolutely nothing wrong. The only trouble is that a lot of us like myself couldn't handle it.
Why do I say that? Because when I start to accumulate, the more I have, the more I want. The more I wanted, the more obsessed I became. Like what I showed you earlier on, all I can was basically to get more possessions, to reach the pinnacle of what society did to us, of what society wants us to be. I became so obsessed that nothing else really mattered to me. Patients were just a source of income, and I tried to squeeze every single cent out of these patients.
A lot of times we forget, whom we are supposed to be serving. We become so lost that we serve nobody else but just ourselves. That was what happened to me. Whether it is in the medical, the dental fraternity, I can tell you, right now in the private practice, sometimes we just advise patients on treatment that is not indicated. Grey areas. And even though it is not necessary, we kind of advocate it. Even at this point, I know who are my friends and who genuinely cared for me and who are the ones who try to make money out of me by selling me "hope". We kind of lose our moral compass along the way. Because we just want to make money.
Worse, I can tell you, over the last few years, we bad mouth our fellow colleagues, our fellow competitors in the industry. We have no qualms about it. So if we can put them down to give ourselves an advantage, we do it. And that's what happening right now, medical, dental everywhere. My challenge to you is not to lose that moral compass. I learnt it the hard way, I hope you don't ever have to do it.
Secondly, a lot of us will start to get numb to our patients as we start to practise. Whether is it government hospitals, private practice, I can tell you when I was in the hospital, with stacks of patient folders, I can't wait to get rid of those folders as soon as possible; I can't wait to get patients out of my consultation room as soon as possible because there is just so many, and that's a reality. Because it becomes a job, a very routine job. And this is just part of it. Do I truly know how the patient feels back then? No, I don't. The fears and anxiety and all, do I truly understand what they are going through? I don't, not until when this happens to me and I think that is one of the biggest flaws in our system.
We’re being trained to be healthcare providers, professional, and all and yet we don't know how exactly they feel. I'm not asking you to get involved emotionally, I don't think that is professional but do we actually make a real effort to understand their pain and all? Most of us won’t, alright, I can assure you. So don't lose it, my challenge to you is to always be able to put yourself in your patient's shoes.
Because the pain, the anxiety, the fear are very real even though it's not real to you, it's real to them. So don't lose it and you know, right now I'm in the midst of my 5th cycle of my chemotherapy. I can tell you it’s a terrible feeling. Chemotherapy is one of those things that you don't wish even your enemies to go through because it's just suffering, lousy feeling, throwing out, you don't even know if you can retain your meals or not. Terrible feeling! And even with whatever little energy now I have, I try to reach out to other cancer patients because I truly understand what pain and suffering is like. But it's kind of little too late and too little.
You guys have a bright future ahead of you with all the resource and energy, so I’m going to challenge you to go beyond your immediate patients. To understand that there are people out there who are truly in pain, truly in hardship. Don’t get the idea that only poor people suffer. It is not true. A lot of these poor people do not have much in the first place, they are easily contented. for all you know they are happier than you and me but there are out there, people who are suffering mentally, physically, hardship, emotionally, financially and so on and so forth, and they are real. We choose to ignore them or we just don't want to know that they exist.
So do think about it alright, even as you go on to become professionals and dental surgeons and all. That you can reach out to these people who are in need. Whatever you do can make a large difference to them. I'm now at the receiving end so I know how it feels, someone who genuinely care for you, encourage and all. It makes a lot of difference to me. That’s what happens after treatment. I had a treatment recently, but I’ll leave this for another day. A lot of things happened along the way, that's why I am still able to talk to you today.
I'll just end of with this quote here, it's from this book called Tuesdays with Morris, and some of you may have read it. Everyone knows that they are going to die; every one of us knows that. The truth is, none of us believe it because if we did, we will do things differently. When I faced death, when I had to, I stripped myself off all stuff totally and I focused only on what is essential. The irony is that a lot of times, only when we learn how to die then we learn how to live. I know it sounds very morbid for this morning but it's the truth, this is what I’m going through.
Don’t let society tell you how to live. Don’t let the media tell you what you're supposed to do. Those things happened to me. And I led this life thinking that these are going to bring me happiness. I hope that you will think about it and decide for yourself how you want to live your own life. Not according to what other people tell you to do, and you have to decide whether you want to serve yourself, whether you are going to make a difference in somebody else's life. Because true happiness doesn't come from serving yourself. I thought it was but it didn't turn out that way. With that I thank you, if you have any questions you have for me, please feel free. Thank you.
-
Aleoca333 asked :
Does Gibbs free energy have anything to do with shifting of POE (position of equilibrium)? A negative G means POE lies very much to the right of reaction?
Yes that's right.But beware the difference between delta G (Gibbs free energy) and delta G-standard (standard Gibbs free energy).
Delta G-standard = - n x F x cell potential
and Delta G-standard = - R x T x ln K
Notice that in both formulae above, we can relate standard Gibbs free energy of any reaction, to it's equilibrium constant (K or Kc or Kp) and to its cell potential (cell potential = reduction potential @ cathode + oxidation potential @ anode).
When cell potential is positive, we say the reaction is thermodynamically feasible and spontaneous, and the current flows in the predicted direction from our designated anode to our designated cathode. The more positive the cell potential, the more the position of equilibrium lies to the right. The more negative the cell potential, the more the position of equilibrium lies to the left.
When delta G-standard < 0, then Kc > 1 and the reaction is product favoured.
When delta G-standard = 0, then Kc = 1 and the reaction is neither reactant nor product favoured.
When delta G-standard > 0, then Kc < 1 and the reaction is reactant favoured.
Aleoca333 replied :
Alright thanks Ultima. Anyway, for chem planning, can we use a divided flask, for a reaction between a liquid/aqueous and a solid? Meaning liquid one side solid side.
Yes, that's certainly acceptable, as long as you clearly illustrate, label, annotate and explain how you would use it (ie. tilt the flask to mix the reagents to begin the reaction).
Divided Flask :
https://www.google.com.sg/search?q=divided+flask -
Balance the following equation :
Cu + HNO3 -> Cu(NO3)2 + H2O + NO
Solution :
Observe that the OS (Oxidation State) of copper increases from 0 to +2, while the OS of nitrogen decreases from +5 (in the nitrate(V) ion) to +2 (in nitrogen monoxide aka nitric oxide). Hence to balance redox equations, identify and write out both half-equations.
Oxidation half-equation :
Cu ---> Cu2+ + 2e-
Reduction half-equation :
4H+ NO3- + 3e- ---> NO + 2H2O
Balanced Redox :
Multiplying by lowest common multiple to balance electrons,
[O]x3 + [R]x2
=
3Cu ---> 3Cu2+ + 6e-
+
8H+ + 2NO3- + 6e- ---> 2NO + 4H2O
=
3Cu + 8H+ + 2NO3- ---> 3Cu2+ + 2NO + 4H2O
Adding the 6 NO3- counter ions on both sides to convert the ionic equation into the chemical equation :
3Cu + 8HNO3- ---> 3Cu(NO3)2 + 2NO + 4H2O -
Memo asked :CoCl2 is the catalyst I can understand --> mainly got the idea when pink appears again.
But how is tartaric acid a ligand? (my guess - the lone pair of e- at O)
Perhaps, I would like to understand what is going on here...UltimaOnline replied :
The lone pair and negative formal charge on the O atom of the tartarate (a carboxylate) ion allows it to function as a ligand, which serves the important function of stabilizing the higher +3 OS of cobalt, thereby making the catalyzed redox reactions (in which Co2+ is oxidized to Co3+ and H2O2 reduced to H2O, and Co3+ is reduced to Co2+ and H2O2 is oxidized to O2) thermodynamically feasible under standard conditions.
The stronger the ligand, the stronger the coordinate dative bond, the more stable the metal ion (of the coordination complex), the more positive (or less negative) the oxidation potential, the more positive the cell potential, the more the position of equilibrium lies to the right, the more negative the Gibbs free energy, the more thermodynamically feasible the reaction. -
^_^
-
^_^
-
kickme asked :
Just to clarify for #1, you are referring to the carbon atom when u write about the positive charge, right? And what is the difference between partial/formal/ionic positive charge?
From #1, is it correct to say that the greater the partial positive charge on the carbon atom of an alkyl halide, the more reactive the alkyl halide is?
Yes of course (to both your questions).As an example, lets look at aldehydes and ketones.
The carbonyl C atom of ketones and aldehydes have a partial positive charge, due to both induction (O being more electronegative than C, consequently the resonance hybrid has a partial positive charge on the C atom, and a partial negative charge on the O atom) as well as by resonance (the pi bond can delocalize onto the O atom as a lone pair, resulting in a resonance contributor which has a unipositive formal charge on the C atom and a uninegative formal charge on the O atom, consequently the resonance hybrid has a partial positive charge on the C atom, and a partial negative charge on the O atom). Hence the electrophilic atom is the carbonyl C atom, within the electrophile molecule of ketones and aldehydes.
In CarboCations (ie. C atom with a 3 bond pairs, 0 lone pairs; 3 + 0 = 3 but since C is in Group IV, hence) the C atom has a unipositive formal charge.
In CarbAnions (ie. C atom with a 3 bond pairs, 1 lone pair; 3 + 2 =5 but since C is in Group IV, hence) the C atom has a uninegative formal charge.
The conjugate base of water, the (uninegative ionic charged) hydroxide ion, has a uninegative formal charge on the O atom (ie. O atom with 1 bond pair, 3 lone pairs; 1 + 6 = 7 but since O is in Group VI, hence a uninegative formal charge).
The conjugate acid of water, the (unipositive ionic charged) hydroxonium ion, has a unipositive formal charge on the O atom (ie. O atom with 3 bond pairs, 1 lone pair; 3 + 2 = 5 but since O is in Group VI, hence a unipositive formal charge).Ionic charge is the sum of formal charges.
The fully protonated conjugate acid form of a diprotic diamine (ie. a molecule with 2 amine groups) has 2 N atoms with a unipositive formal charge, hence the ionic charge for this species, is dipositive (ie. 2+).
The fully deprotonated conjugate base form of a diprotic dicarboxylic acid (ie. a molecule with 2 carboxylic acid groups) has 2 O atoms with a uninegative formal charge, hence the ionic charge for this species, is dinegative (ie. 2-).
-
Kickme asked :
Question 1: In order to prevent the oxidation of chloroform, it is kept in dark bottles. What does this suggest about the nature of this oxidation?
I have 2 considerations: 1) It is an endothermic reaction as it requires the supply of energy from light
2) it is a free radical substitution reaction as it requires UV light
Question 2: Also another question I have is from 2007 'A' Paper 2 qn 2 which asks: What is unusual about this reaction?
And the answer is it is a disproportionation/redox reaction. Is that even unusual?? I answered that Cl2 is acting as a reducing agent.UltimaOnline replied :
The mechanism for the oxidation of chloroform into phosgene does indeed involve free radicals (it is technically incorrect to say free radical substitution, as it is a free radical oxidation reaction; it is safest to simply say "the reaction mechanism involves free radicals").
https://en.wikipedia.org/wiki/Cannizzaro_reaction
It is also incorrect to say that the reaction must therefore be endothermic, because even exothermic reactions require activation energy to initiate.
The Cannizzaro reaction is indeed unusual, and quite fascinating, beautiful and elegant; because (while inorganic chemistry disproportionation reactions are a dime a dozen) the Cannizzaro reaction is one of very few organic chemistry disproportionation reactions, in which an aldehyde (+1 OS of the C atom) is simultaneously reduced into its primary alcohol form (-2 OS of the C atom) , and also oxidized into it's carboxylic acid form (+3 OS of the C atom).
2 Aldehyde ---> 1 Alcohol + 1 Carboxylic Acid
The rather elegant Cannizzaro mechanism :
-
InfinityFrost asked :
TJC/2012; Paper 2, Q.5(b)
I have two answers for this. But I'm not sure which is more acceptable (if this is the right term to use). Or if there's any mistake in my working. I remember my method working fine for all the questions I've done. And I think this is an issue of approximation.
An aqueous solution of lactic acid of concentration 0.100 moldm^-3 has a pH value of 2.04. Calculate the value of the dissociation constant.
My answer
Lactic acid dissociates to give oxonium ions + conjugate base
Ka = [(10^-2.04)^2]/[0.100 - 10^-2.04) = 9.15 x 10^-4
Answer key
Ka = [(10^-2.04)^2]/[0.100) = 8.32 x 10^-4
Your answer of
Ka = [(10^-2.04)^2]/[0.100 - 10^-2.04) = 9.15 x 10^-4
is more accurate compared to TJC's answer key of
Ka = [(10^-2.04)^2]/[0.100) = 8.32 x 10^-4
While Cambridge usually accepts both answers, but (depending on Mark Scheme guidelines which changes slightly from paper to paper) the less accurate TJC method may be penalized.
This is because, for the approximation of equilibrium molarity back to initial molarity to be mathematically accurate up to 3 significant figures, the initial molarity of the species undergoing the reaction must be over 1000 (notice the 3 zeros, since accuracy is required up to 3 significant figures) times larger compared to the Kc (eg. Ka or Kb) value.
The exam-smart thing to do, when you're not sure which is the better answer to give, is to give both answers, but with qualification and/or explanation.
(If you do not qualify or explain, then the examiner would think you're trying to cheat by giving two different answers, and of course you'll be penalized.)
For instance, if the question does not specify "hot" or "cold" when KMnO4 is reacted with a reactant molecule that could generate different possible products (eg. with an alkene double bond, benzene ring alkyl substituent, primary alcohol, etc), the exam-smart thing to do is to give both (or all) possible answers, but with qualification and explanation.
"If heated under reflux with concentrated KMnO4, then the product is..."
"If reacted with cold, dilute KMnO4, then the product is..."
Note that the concentration of KMnO4, and the pH of the solution, is of secondary importance to the reaction temperature, in determining the extent of oxidation by KMnO4. -
You need to be able to draw out the Reflux Apparatus for Planning :
http://www.rod.beavon.clara.net/reflux.htm
In most cases (eg. most organic chemistry reactions), either just "heat" or the more thorough "heat under reflux" (I don't recommend ever just writing "reflux" alone) are both acceptable by Cambridge.
In some cases (eg. oxidation of primary alcohol to aldehyde only), you must write "heat and distill" (and K2Cr2O7, not KMnO4, must be used). "Heat under reflux" would be penalized.
For the Iodoform test, it is best to write "warm", as "heat" would provide excessive activation energy for further hydrolysis of the iodoform (ie. triiodomethane) and the precipitate would dissolve. Here, "heat" may or may not be penalized, but "heat under reflux" would definitely be penalized.
In synthesizing some organic compounds (eg. nitrobenzene) which require high activation energies, prolonged heating is required, and therefore "heating under reflux" and not just "heat", must be written. And if a particular temperature is required, that must also be written in (eg. > 60 deg C to obtain dinitrobenzene).
However, in some other industrial organic synthesis, due to the complex industrial machinery setup used (in which reflux apparatus is not appropriate and possibly even counter productive), only a particular temperature and catalyst must be specified, and "heat under reflux" should not be written.
H2 Chem students are required by the syllabus to memorize the large-scale industrial synthesis methods, which means that although in a small lab on a small scale synthesis, "heating under reflux" may be appropriate, but if the large-scale industrial method does not use "reflux", then you should not include "reflux" in your description. Or vice-versa.
Examples of such industrial synthesis methods are the dehydration of alcohols to alkenes (excess concentrated H2SO4, heat at 170 deg C), and the hydration of alkenes to alcohols (heat with steam at 300-330 deg C and 60-70 atm, with H3PO4 catalyst). "Reflux" must not be written for these reactions.
Generally, if asked to describe a simple test to distinguish between two organic compounds, in which the test is to be carried out on a small scale in test tubes, then "heat" must be written, and not "heat under reflux". Because that would require reflux apparatus, which cannot be connected to a test tube, and which would not constitute a "simple" test, and would be a (Cambridge) frowned-upon waste of resources.
In contrast, in answering a "design a synthesis pathway" question, to have a reasonably good yield of the product at equilibrium, especially if one or more of the reactants are volatile (ie. low boiling point), then "heat under reflux" must be written, and not just "heat" alone. Low yield syntheses as a result of lazy failure to use reflux apparatus, are a (Cambridge) frowned-upon waste of resources.
When in doubt (don't trust Singapore JC notes, as all 20 different JCs have significant differences in their notes across all topics), you should use reliable sources of H2 Chem study material to memorize the syllabus required descriptions, including when to write "heat" and when to write "heat under reflux". -
Kickme asked :
TYS 2010, Paper 3, question 5(a)
At the anode, 2 reactions are occuring, Cu -->Cu2+ and Zn -->Zn2+
This is a simultaneous discharge.
While at the cathode, only Cu2+ --> Cu occurs...
I agree that Cu2+ is reduced at the cathode. However, at the anode 2 reactions take place, Cu --> Cu2+ and Zn --> Zn2+
Should Zn be preferentially oxidised? Of which, will result in no available Cu2+ to be reduced at the cathode.First of all, don't (mis)use the term "discharge", which means "to remove a charge". Hence, while you can say Cu2+ ions are discharged as they are reduced to Cu at the cathode, you cannot say Cu atoms are being "discharged" as they are oxidized to Cu2+ at the anode, because they are now attaining a charge, not losing a charge.
Heck, just totally don't use the word "discharge" at all. Seriously.
Cambridge prefers you to phrase the answer as follows (notice there is no "discharge" in the answer) :
At the anode, copper metal is being oxidized since most of the anode itself is made up of copper. Cu(s) ---> Cu2+(aq) + 2e-. (There are two reasons why you can't just oxidize only the zinc, and not oxidize the copper at all. Firstly, not all the zinc particles are on the exterior of the anode, some zinc particles are trapped in the interior of the anode and cannot be oxidized until the outer copper layers have oxidized away into the electrolyte solution. Secondly, the power supply is forcing the flow of electrons, which means the anode has no choice but to oxidize away whatever is available, or else the current can no longer flow and human civilization would be destroyed. There simply isn't enough zinc impurities to keep the current flowing at the rate determed by the power supply. So copper has no choice to but be oxidized en masse, together with the tiny bits of zinc, to keep the current flowing).
Little bits of metallic zinc impurities within the anode are also oxidized, since the oxidation potential of Zn to Zn2+ is more positive compared to the oxidation potential of Cu to Cu2+. Bits of metallic silver impurities at the anode, are not oxidized, because the oxidation potential of Ag to Ag+ is less positive compared to the oxidation potential of Cu to Cu2+.
While both Cu2+(aq) and Zn2+(aq) ions both migrate towards the cathode, however because the reduction potential of Cu2+ to Cu is significantly more positive, compared to the reduction potential of Zn2+ to Zn, hence the Cu2+ ions are preferentially reduced at the cathode, leaving the Zn2+ ions in the aqueous state in the electrolyte solution.
Consequently, the cathode now consists of pure copper, without either the less reactive silver impurities, or the more reactive zinc impurities, that were initially present at the anode. -
Kickme asked :
Is it correct to say:
During denaturation, the quaternary, tertiary and secondary structures will be altered as the weak interactions maintaining these structures together such as Van der Waals’ forces of attraction, hydrogen bonding, ionic bonding and disulfide linkages are overcome.
Yes it's ok, but don't include "disulfide linkages" if you wanna use the word "weak" interactions. Coz disulfide bonds, being covalent, are stronger than the other R group interactions, and a protein is considered denatured as long as some of these R group interactions are disrupted leading to loss of the protein's original biological function.
Hence, a better definition of denaturation is :
Denaturation refers to the loss of a protein's intended biological function or activity, due to the disruption and loss of its original secondary and/or tertiary structures, as a result of any of the following : high temperatures, extreme pH, heavy metal ions, mechanical agitation, non-aqueous protic solvents.
If the question asks you in more detail about how each R group interaction is disrupted, you will need to go into more detailed explanations, diagrams or equations, eg. for how heavy metal ions disrupt ionic and disulfide bonds/bridges/linkages.
-
Kickme asked :
Why do you not include quaternary structures in denaturation? Also, my school notes doesn't provide any chemical equations under how metal ions disrupt ionic and disulfide linkages.
Not all proteins have quarternary structures (in fact, most don't). Cambridge doesn't care whether you include "quarternary structure", as long as you do mention "tertiary structure" in your answer on denaturation.
Google be thy sword, Wikipedia by thy shield. Chemical equations that illustrate how heavy metal ions disrupt ionic bonds and disulfide linkages are required in the H2 syllabus.
-
Kickme posted :
Can I ask, how do i balance the chemical equation for Fehling's and Tollen's reagent with Na+?
UltimaOnline replied :With great difficulty. Ok seriously, these two are equations that will be much easier to handle by just memorizing the coefficients. Balancing them manually is too time consuming. Also, the chance that Cambridge will ask you to write out these balanced equations is not high, so if you find it difficult to memorize or balance, just skip this part.
Kickme posted :
However, the equation given in my notes is that of only OH-, without Na+, and I don't see how I can add the same no. of Na+ on both sides.
Take tollen's reagent for example,
{RCHO} + 2{[Ag(NH3)2]+} + 3{OH-} --> 2Ag + RCOO- + 4NH3 + 2H2O
If I insist on balancing with Na+, I will end up with 3 Na+ on the left side and just 1 Na+ on the right side... And furthermore, the charges will not balance
Just to add, is it alright to write tollen's reagent if the question asks to suggest a simple test or to synthesis a product?UltimaOnline replied :
You have difficulty balancing the equation when you add the spectator counter ions, because you're considering the Na+ counter cations; there is actually one other counter ion present - the NO3- counter anion (the coordination compound in Tollens' reagent is actually diamminesilver(I) nitrate).
To save yourself trouble, leave out the spectator counter ions, and only write in the participating ions.
FYI, the RJC Organic Chem book does teach students step-by-step how to balance both the Fehling's and Tollens' equations.
Yes, Cambridge accepts the name "Tollens' reagent", you do not need to write out the name or formula of the diamminesilver(I) nitrate active species present. Same for Fehling's, and 2,4-DNPH (ie. abbreviation acceptable).blabla123 posted :
Ahhh seriously?! I think the school very kiasu.. As in we were told strictly to say exactly ammonicial solution of agnh3 2+ containing agno3 and alkaline solution of copper ii tartrate. Oh and 24dnph must spell in full cannot short form. Well at least i know its not necesssary but in case during exams i forgot the compounds or smth i can just right the name of the reagent or DNPH less i write wrongly and get penalised. Thx!
UltimaOnline replied :
Silly school, sabotaging you students with counter-productive answers. That's why I always advise, don't over-trust in Singapore schools, nor over-rely on their notes.
Three reasons why your school's instructions are silly (and downright risky) :
1) You're wasting valuable time under exam conditions, writing out unnecessarily long answers.
2) You're annoying the Cambridge marker (didn't your school teach that it's bad manners to annoy someone who's holding your 'A' level grades in his/her hands), because now he/she has to do more work to check your spelling and formulae, when all he/she wanted was a short and sweet "Tollens' reagent", so he/she can complete his/her marking workload sooner and return home earlier to a nice dinner with his/her loved ones.3) If you make a typo error (eg. for names such as bistartratocuprate(II)*, or formula such as [Ag(NH3)2]+, etc), you will be penalized.
* Your school committed a grevious error, Fehling's solution isn't copper(II) tartrate. The active species in Fehling's solution is the coordination compound disodium dipotassium bistartratocuprate(II). See my point?
-
Kickme asked :
Why is it Tollens' and not Tollen's or Tollens's?

Because this gentlemen is Mr Tollens, not Mr Tollen (and Tollens's is just bad grammar). Mispelling his name constitutes an unacceptable disrespect towards a pioneering Chemist senior, and incur will Cambridge's outrage and you will be penalized 10 marks for it (minus 1 mark for spelling error, minus 9 marks for disrespect to a Chemist senior).Just kidding lah, Cambridge will accept your answer whether you write it correctly as Tollens', or incorrectly as Tollen's, but writing it as Tollens's is pushing it a bit.
-
BlackToast asked :
1. do u write phosphourous as P or P4 in the standard state?
2. what if for example they give u an alkyl halide and say NAOH is added to it, but no heat. will nucleophilic substitution still occur?Q1.
For the purpose of Inorganic Chem - Periodicity, eg. explaining trend of Melting Points across Period 3, you have to write them as S8, P4, Cl2, Ar, etc. This will enable you to explain why melting point decreases across the period :
The greater the number of electrons present, and the larger the molecular size, hence the more polarizable the electron charge clouds, hence the greater the magnitude of (instantaneous and induced) dipoles and partial charges, hence the stronger the electrostatic attractions, and hence the stronger the intermolecular van der Waals forces of attraction (required to be overcome), and therefore the higher the melting and/or boiling point.
Q2.
At room temperature, the reaction will occur, but very slowly (ie. thermodynamically feasible, but kinetically non-feasible). Hence, for synthesis questions, you must specify "heat under reflux" for hydrolysis to occur at an acceptable rate. Heating speeds up the reaction by increasing the percentage of reactant particles with energy equals or exceeding the required activation energy.
Nonetheless, on a related note, consider this : upon mixing AgNO3(aq) and alkyl halides (dissolved in ethanol), nucleophilic substitution will occur at a significant rate at room temperature, even without heating.
This is due to the exothermic (hence thermodynamically favourable) coupling of the Ag+ silver ion with the X- halide ion, to generate the precipitate, which not only provides the activation energy required (hence also kinetically feasible), but also (as predicted by Le Chatelier's principle) shifts the position of equilibrium over to the right.
In fact, the time taken for the silver halide precipitate to be observed, is an important test in the H2 syllabus to differentiate between acyl halides and alkyl halides, and between the different akyl halides. (Notice that acyl and alkyl fluorides and generally fluorine compounds, behave exceptionally from the other halogens/halides, and are hence exempted from the H2 syllabus).
The candidate needs to be able to explain this in terms of covalent bond dissociation enthalpies, in turn due to the effectiveness of (head-on or end-on) orbital overlap to form the sigma bond, in turn due to the varying diffuseness of the varying valence quantum shell orbital used. -
Prototype asked :
I'm having some misconception about the hybridisation of H2O
H2O has sp3 hybrid orbitals is it because that it does not have any pi bonds from the central atom to the terminal atoms?For orbital hybridization, look at the electron geometry, not the molecular or ionic geometry.
(A further example would be ammonia : with 1 lone pair and 3 bond pairs, the electron geometry is tetrahedral and the molecular geometry is trigonal pyramidal. Last example : the uninegative triiodide ion, I3-, has 3 lone pairs and 2 bond pairs on the central I atom, hence the electron geometry is trigonal bipyramidal, and the ionic geometry is linear, since lone pairs occupy the equatorial positions for trigonal bipyramidal electron geometry; but note that lone pairs occupy the axial positions for octahedral electron geometry.)
In water, the valence shell of the O atom has 2 lone pairs and 2 bond pairs. Hence, with 4 valence electron charge clouds, its electron geometry would be tetrahedral, as predicted by VSEPR theory (whose objective is to maximize stabilities by minimizing electron pair repulsions).
If the O atom were to utilize its unhybridized orthogonal p orbitals to form its sigma bonds (via a head-on or end-on overlap with the s orbital of H), the H2O bond angles would be likewise orthogonal (ie. 90 deg). This would not be optimally stable, due to (guys only like girls, guys don't like guys) significant bond pair - bond pair repulsion.
Hence, in order to achieve the ideal tetrahedral bond angles, the O atom hybridizes its orbitals to achieve 4 equivalent sp3 orbitals which are 109.5 deg away from each other (assuming the guys are heterosexual, guys need a healthy amount of personal space between them and other guys, to maintain social stability). Of these four sp3 orbitals, 2 of them are fully filled (ie. lone pairs), and 2 of them are half-filled, which are therefore used to overlap head-on or end-on, with the s orbitals of the H atoms, to form the two sigma bonds in H2O. -
^_^
-
^_^